










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr vol.90 no.2 Ciudad de la Habana abr.-jun. 2018
PRESENTACIÓN DE CASO
Incontinencia pigmenti y manifestaciones neurológicas
Incontinentia pigmenti and neurological manifestations
Ernesto Portuondo Barbarrosa, Pilar María Acuña Guilarte, Ana Gloria González Bécquer, María Caridad Rigautdi, Iraida de la Caridad Pérez Ferrer
Hospital Pediátrico Docente Centro Habana. La Habana, Cuba.
RESUMEN
Introducción: la incotinencia pigmenti es una genodermatosis rara ligada al cromosoma X, afecta al sexo femenino y tiene diferentes expresiones clínicas en una misma familia.
Presentación del caso: preescolar de 20 meses de edad, con antecedente familiar de incotinencia pigmenti, que presentó lesiones típicas en la piel desde la primera semana de vida, de aspectos lineales, vesículo-costro-ampollosas, verrucosas, y luego hiperpigmentadas, en diferentes fases y múltiples brotes. Comienza desde el primer mes de vida con crisis epilépticas que evoluciona a una encefalopatía de West, con buena respuesta a la vigabatrina y control de los espasmos infantiles.
Conclusiones: la incotinencia pigmenti se caracteriza por afectar, de forma variable, a los tejidos derivados del neuroectodermo, la piel y otras faneras, ojos y el sistema nervioso central, provoca daño multisistémico. Las lesiones de la piel son las más significativas desde el nacimiento, y la biopsia de piel confirma el diagnóstico.
Palabras clave: incontinencia pigmenti; genodermatosis ligada al cromosoma X; encefalopatía epiléptica (síndrome de West).
ABSTRACT
Introduction: incotinentia pigmenti is a rare genodermatosis linked to the X chromosome. It affects the female sex and has different clinical manifestations in the same family.
Case presentation: a 20-month-old infant with a family history of incotinentia pigmenti, who from the first week of life presented typical lesions on the skin of linear, vesicular-crust-bullous, warty, and then hyperpigmented aspects, in different phases and multiple outbreaks. From the first month of life, the patient presented epileptic seizures that evolved to West encephalopathy, with good response to vigabatrin and control of infantile spasms.
Conclusions: incotinentia pigmenti is characterized by affecting, in a variable way, the tissues derived from the neuroectoderm, the skin and other skin´s structures, the eyes and the central nervous system causing multisystem damage. Skin lesions are the most significant since birth, and skin biopsy confirms the diagnosis.
Keywords: incontinentia pigmenti; genodermatosis linked to the X chromosome; epileptic encephalopathy (West syndrome).
INTRODUCCIÓN
La incontinencia pigmenti (IP) es una genodermatosis de escasa frecuencia, ligada en forma dominante al cromosoma X. Las manifestaciones cutáneas están siempre presentes, y se dividen en 4 fases.1-5 Afecta la pigmentación de la piel, y suele estar asociada con una gran variabilidad de alteraciones en los ojos, las uñas, el pelo, los dientes, el esqueleto, el corazón y el sistema nervioso central.1-8
Se ha descrito y observado una expresión clínica muy variable en miembros de una misma familia. Algunas pacientes solo presentan síntomas y signos cutáneos, mientras que otras pueden sufrir hasta una severa incapacidad neurológica y/o oftalmológica.1-6
El objetivo es reportar un caso clínico de una paciente que recibimos a los 46 días de vida, derivada desde el pediatra familiar, por presentar desde su nacimiento un cuadro cutáneo de vesículas y ampollas con contenido seroso, que sospechaban infección estafilocócica, asociada, a posteriori, a otras manifestaciones clínicas neurológicas.
El aspecto de las lesiones, su distribución y su evolución, además del antecedente materno, de la abuela y tía abuela de lesiones cutáneas similares en la infancia, nos permitió establecer el diagnóstico de IP, con colaboración del servicio de Dermatología y Anatomía patológica. Se resaltan las tres primeras fases cutáneas de esta enfermedad en la niña y las manifestaciones neurológicas, por lo que constituye un caso de IP familiar.
PRESENTACIÓN DEL CASO
Preescolar de 24 meses de edad actual, nacida de padres jóvenes, no consanguíneos, de parto distócico a las 38,5 semanas por afección materna (meconio y extracción difícil), con peso de riesgo (2 960 g) y Apgar 9/9.
La madre presentó serología no/reactiva, negativo a VIH y diabetes gestacional, y por esta última razón, se le realizan complementarios al nacer la niña (glucemia 4,7 mmol/L, hematocrito 0,55 y calcio normal).
A las 59 horas de nacida comenzó a presentar lesiones en la piel vesículo-papulo-ampollo-pustulosas, con contenido seroso y micelerico, diseminadas en el tronco, ya predominio de las extremidades, diagnosticadas en un inicio como piodermitis, para lo cual se le aplicó tratamiento tópico con hibitane acuoso y gentamicina. Por este cuadro se le ingresa en el servicio de Neonatología para mejor seguimiento. A los 4 días es valorada nuevamente porque no había mejoría de las lesiones en piel, y se diagnostica eritema múltiple menor y/o un eritema tóxico de recién nacido. Se le realiza perfil de sepsis, y todo se presenta dentro de límites normales.
En el seguimiento se notó resolución de las lesiones de tronco, no así de las de las extremidades, que evolucionaron de forma polimorfa hacia lesiones pigmentadas, decamativas y vesiculo-postulosas, tanto en miembros superiores e inferiores, y espontáneamente a aspecto verrucosa (figura 1 A). Es en este momento cuando se le hace el diagnóstico de IP, teniendo en cuenta la clínica y los antecedentes familiares de estigma de esta enfermedad (madre, abuela y tía abuela materna), aunque no estudiadas completamente en su momento.
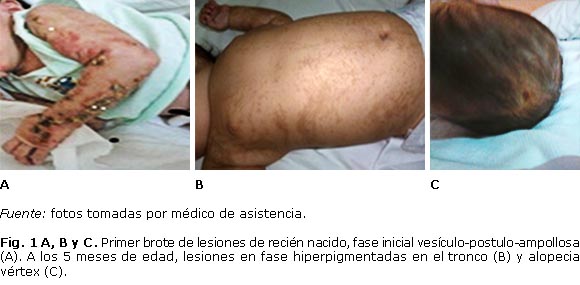
En los primeros 3 meses de vida presentó varios brotes de lesiones, sobretodo, en las extremidades, casi siempre de disposición lineal, siguiendo las líneas de Blaschko. Luego evolucionaron hacia lesiones verrucosas, y más adelante se hicieron hiperpigmentadas, abarcando ahora, además de las extremidades, el tronco y la cara (siempre siguiendo las líneas de Blaschko), en forma de remolino asociada a alopecia vértex (figura 1 B y 1 C). Los brotes continuaron hasta después de cumplir el primer año.
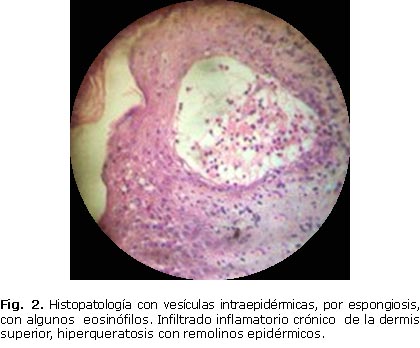
Histopatología de piel lesionada (figura 2): se observan vesículas intraepidérmicas, por espongiosis, con algunos eosinófilos. Infiltrado inflamatorio crónico de la dermis superior, e hiperqueratosis con remolinos epidérmicos. Como está en un estadio avanzado de la enfermedad, muestra melanófagos cargados de melanina y degeneración hidrópica de la basal.
A los 58 días comenzó con movimientos clónicos de miembro superior izquierdo, con desviación de la mirada hacia arriba, desconexión y progresaba en marcha, llegando a presentar un estado epiléptico convulsivo focal motor discognitivo, con alteración del estado de alerta, evolución a bilateral clónico. Ingresó en la Unidad de Cuidados Intensivos Pediátricos (UTIP) durante 48 horas, y se constató crisis electro-clínica en electroencefalograma (EEG), crisis focal motora clónica facial, con lateralización de la cabeza a derecha, desviación de la mirada, clonías palpebrales y en marcha a miembro superior derecho y luego izquierdo.
El EEG inicia con electro decremento fronto-centro-parietal izquierdo, seguido con actividad rápida, que aumenta en amplitud y frecuencia, se propaga a región homóloga contralateral, y termina con enlentecimiento global con ondas delta. Se inicia tratamiento con fenobarbital endovenoso con resolución del estado de mal epiléptico focal, y luego continuo por vía oral. A los 4 meses y 16 días comenzó con otros eventos epilépticos, que consistieron en espasmos infantiles (EI) en salva o clúster, de flexión y mixtos, un mínimo de 45 y máximo 110 EI en 24 horas. Se confirma con EEG, que constata un trazado con hipsarritmia (figura 3), por lo que se diagnostica una encefalopatía epiléptica de síndrome de West (SW), para el cual se inicia tratamiento con vigabatrina (VGB), relacionando crisis focales previas y EI.
Dada la intensidad y frecuencia de estas crisis, se asoció tratamiento con hormona adenocorticotrópica sintética (ACTH). Recibió dosis en días alternos durante 2 semanas, y dosis única semanal durante 2 semanas (2 décimas intramuscular [IM]/1 mg=1 mL) sin reacciones adversas, combinada con VGB. Desaparecen los EI en los primeros 2 meses del tratamiento, y la hipsarritmia en el EEG. Con dosis de VGB de 1 500 mg (1½ tableta c/12 horas), tiene un control total de las crisis. Después de 12 meses sin EI y EEG con privación de sueño, con trazado de base organizado y espigas multifocales, se sustituyó la VGB por valproato de sodio, a dosis de 25 mg/kg/día, sin evidencia de crisis epilépticas.
El desarrollo psicomotor (DPM) de la paciente estuvo comprometido con un retardo de más o menos 4 meses. A los 6 meses, no tenía sostén cefálico completo, discreta hipotonía axial, mayor movilidad en hemicuerpo derecho (cuadriparesia hipotónica con doble hemiparesia a predominio izquierdo, no sedestación, no rola ni gira sobre su abdomen, y no intercambia objetos de una mano a la otra), el gorjeo y la sonrisa son escasos. Logró la sedestación y rolar sola entre los 10 y 11 meses.
Desde el punto de vista oftalmológico se valoró inicialmente, y se constató una posible atrofia del nervio óptico, en curso con la consecuente disminución de la agudeza visual. Clínicamente, presenta una microftalmia, hipertelorismo y estrabismo, con papila pálida e hipoplasia del iris y anomalía de los vasos de la retina, con mayor severidad del ojo derecho, corroborada por servicio de Oftalmología. Tiene retraso dentario: primeros incisivos mediales inferiores a los 10 meses; y actualmente, solo incisivos superiores e inferiores, caninos y primeros molares superiores con hipodontia. No hay evidencia de síntomas y signos cardiacos, y el ecocardiograma no arroja alteraciones estructurales.
Actualmente, y después del control de los EI y sin hipsarritmia en el EEG, tiene mejoría del DPM en la esfera motora gruesa, bipedestación con apoyo a los 14 meses, primeros pasos con apoyo a los 19 meses, y después de los 22 meses da pasos sin apoyo. En la esfera motora fina, la pinza digital la logra a los 12 meses, y a los 20 intenta garabatos. Esto ha sido posible gracias al trabajo coordinado de los padres y los profesionales del área de estimulación precoz y rehabilitación.
En la socialización, obedece órdenes y señala objetos con el dedo, da besos y hace "torticas". El lenguaje fue solo con ruidos hasta los 14 meses, en que comienza los primeros monosílabos, y a los 20 meses dice mamá y papá, pero no frases de dos palabras. No presenta afectación del perímetro cefálico (47 cm/50 percentil para su edad), e hipotonía discreta, con predominio del hemicuerpo izquierdo.
Se realizó tomografía axial computarizada de cráneo simple (TAC) antes del inicio de tratamiento con ACTH, en la cual se evidencian signos de atrofia córtico subcortical fronto, parieto, temporal y bilateral, pendiente de resonancia magnética nuclear de cráneo (RMN). En el EEG actual (24 meses) y con privación sueño, tiene un ritmo de base organizado para su edad, y con espigas multifocales de frecuencia leve. Continúa tratamiento con ácido valproico en monoterapia y sin evidencia de crisis epiléptica.
DISCUSIÓN
La IP, antes conocida como síndrome de Bloch Sulzberger, es una genodermatosis poco frecuente. Fue descrita por Garrod en 1906, y posteriormente definida por Bloch en 1926 y Sulzberger en 1928 como Incontinentia Pigment, citado por Landy,1 Enei2 y Romero y otros,3 basados en los hallazgos histopatológicos característicos -aunque no patognomónicos- de las lesiones cutáneas de la tercera fase de la enfermedad, en la cual se encuentra melanina libre en la dermis, o dentro de los melanófagos dérmicos, elementos descritos en la histopatología de lesiones en la piel de nuestro caso, con lesiones en la tercera fase.4,9
La IP es un trastorno neuroectodérmico sistémico, caracterizado por lesiones cutáneas que evolucionan por estadios: vesiculoso-pustuloso, verrucoso, hiperpigmentado e hipopigmentado. Estas lesiones en la piel desaparecen y evolucionan espontáneamente en el tiempo.1-3,5-10 Se asocia con compromisos oftalmológicos (30 %), neurológicos (5 a 13 %) y odontológicos (30 a 40 %), que son permanentes.6-11 Es una entidad muy rara, que solo ha sido diagnosticada unas 700 veces en todo el mundo, y se presenta casi exclusivamente en mujeres, ya que casi todos los varones mueren in útero y sobreviven si su cariotipo es 47XXY, o si presentan una mutación hipomórfica. Tiene un patrón de herencia autosómico dominante, ligado al cromosoma X dominante (Xq28).1-3,5-11
Las manifestaciones clínicas de la IP son variadas, incluso, en miembros de la misma familia.4 Los hallazgos en la piel generalmente son los primeros en aparecer. La manifestación más frecuente es la alopecia del vértex, que ocurre en el 38 a 50 % de los pacientes.1,7,8
Existe una estrecha relación de la severidad de la entidad por la asociación de manifestaciones oftalmológicas, neurológicas y recurrencias de brotes en las lesiones en la piel.1-11 Dentro de las manifestaciones neurológicas, la microcefalia, la atrofia cerebral, el retraso madurativo con afectación global del neurodesarrollo, la encefalopatía epiléptica relacionada con el SW, la espasticidad, la ataxia y con posterioridad, la discapacidad intelectual, son las más frecuentes, aunque la encefalopatía epiléptica, la encefalomielitis aguda diseminada y la enfermedad cerebrovascular isquémica, son las manifestaciones clínicas más graves.8,11-14 En nuestro caso se describen estas asociaciones de comienzo temprano (antes de los 2 meses), con signos oftalmológicos, neurológicos y odontológicos, por lo que se cumplen los criterios mayores de IP propuestos por Landy y Donnai1 en 1993, y su actualización por Mink y otros15 en 2014.
Los diagnósticos diferenciales que se deberían plantear ante una sospecha de IP van a depender de la etapa en la que se encuentre la enfermedad: para el estadio vesiculoso cabe plantear herpes neonatal, impétigo ampolloso por Staphylococcu saureus, mastocitosis, histiocitosis, epidermólisis ampollosa hereditaria y enfermedad ampollosa por IgA linear. Para el estadio verrucoso, se deben incluir nevus epidérmico linear y liquen estriado. En cuanto al estadio hiperpigmentado, cabe señalar la hipermelanosis nevoide linear y en remolinos, y síndrome de Naegeli-Franceschetti-Jadassohn.1,2
En nuestro caso es válido resaltar -y es significativo- las diferentes expresiones fenotípicas en una misma familia, con mayor severidad en esta tercera generación (caso clínico). La madre, abuela y tía abuela materna solo manifiestan los signos cutáneos y odontológicos (dientes cónicos). El caso que nos ocupa presenta múltiples brotes de lesiones, que se observan en diferentes estadios en cada cita de seguimiento (vesículo-postulosas, verrucosa e hiperpigmentadas), bilaterales, simétricas a predominio de tronco, abdomen y extremidades, similar a lo descrito en otras series,5-7 desde su inicio, a razón de un brote por mes hasta después de cumplir el primer año.
La proporcionalidad de las lesiones cutáneas con el inicio temprano de las crisis epilépticas, desde el primer mes con una epilepsia focal sintomática/estructural-genética por clasificación actual,16-18 que se inició en forma de estado epiléptico focal, y su continuidad con el SW, encefalopatía epiléptica generalizada de la lactancia, con grave afectación en el neurodesarrollo, se describe con alta frecuencia.4-6,8-14 La TAC de cráneo simple arroja atrofia cerebral a predominio frontotemporal, considerado por la nueva propuesta de la Liga Internacional sobre la Epilepsia (ILAE) 2010 y 2017,16-18 de etiología genética, en la que hay afectación multisistémica con posible daño estructural cerebral, y cuyo pronóstico es reservado, por la repercusión de la propia actividad eléctrica cerebral, la posibilidad de crisis epilépticas de difícil control y la propia entidad en sí, con manifestaciones clínicas que involucran el sistema visual, la piel, los dientes y el sistema nervioso central.10-14
Es de resaltar que, a pesar de fenotipo clínico severo, tuvo una buena respuesta al tratamiento con VGB y ACTH combinado, con resolución de los EI y desaparición de la hipsarritmia a los 2 meses de inicio de la medicación, y tiene ya 18 meses libre de EI u otra crisis epiléptica. Se demuestra que, controlar los EI y la actividad epiléptica (hipsarritmia), debe repercutir en una mejoría del DPM, ya en este caso existe la posibilidad de transición a otro síndrome epiléptico, sin evidencia de crisis clínicas. La paciente muestra mejoría en el DPM, en la esfera motora gruesa y fina.
En la mayoría de los pacientes el inicio temprano de múltiples manifestaciones clínicas (antes del primer año), ensombrece el pronóstico, por lo que la detección precoz es importante, y aunque no existe en la actualidad un tratamiento etiológico curativo, el tratamiento es sintomático e interdisciplinario, atendiendo al sistema afectado.1-6,8-12 En nuestro caso se sigue por Neuropediatría, Dermatología, Oftalmología, Fisiatría y Logopedia.
Se concluye que la IP es una genodermatosis rara y poco frecuente, con expresión fenotípica variable en una misma familia, cuya severidad depende de la proporción de los brotes de las lesiones en la piel, las manifestaciones oftalmológicas y el inicio temprano de crisis epilépticas. La afectación es multisistémica y su pronóstico reservado. El diagnóstico debe ser precoz y su tratamiento multidisciplinario. Está muy relacionada con la encefalopatía epiléptica de West (o SW).
Conflicto de intereses
Los autores declaran no tener conflicto de intereses en la realización del estudio.
REFERENCIAS BIBLIOGRÁFICAS
1. Landy SJ, Donnai D. Incontinentia pigmenti (Bloch-Sulzberger syndrome). J Med Genet. 1993;30:53-9.
2. Enei ML, Orellana I, Vargas X, Salazar R, Paschoal F. Incotinencia pigmenti en madre e hija. Relato de caso clínico. Rev Chil Pediatr. 2011;82(3):225-30.
3. Romero A, Tufiño M, Villacís A, Salazar M. Incontinencia pigmentaria o síndrome de Bloch-Sulzberger. Dermatol Rev Mex. 2014;58:539-43.
4. Luna P. Incontinencia pigmenti. Dermatología Pediátrica.Larralde M, Abad E, Luna P, editores. 2ª ed. Barcelona: Ediciones Journal; 2010. p. 65-9.
5. García-Rodríguez Y, Castillo-Maspons G. Incontinencia pigmenti en un recién nacido. Presentación de un caso. Medisur. 2015;13(4): 555-9
6. Inostroza MA, Verdugo FX. Incotinencia pigmentaria asociada a fisura palatina. Reporte de caso. Rev Esp Cir Oral Maxilo Fac. 2013;35(2):74-7.
7. Garcia Garcia A, Hernandez García I, De León Ojeda N, Acosta Sabatés M, Marrón Portales L. Revisión clínica de 28 casos de incotinencia pigmentaria. Rev Cubana Pediatr [serie en Internet]. 2010 [citado 3 de enero de 2017];82(3). Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034-75312010000300003&lng=es
8. Meuwissen MEC, Mancini GMS. Neurologic findings in incontinentia pigmenti: a review. Eur J Med Genet. 2012;55:323-33.
9. Gálvez-Marticorena B, Chambi-Torres J. Incontinentia pigmenti en Cajamarca: Reporte de un caso en lactante. Horizonte Médico. 2015;15(3):57-60.
10. Gonzales Burgos L, Martino Ortiz B, Rodrigo Masi M, Knophelmacher O, Bolla de Lezcano L. Síndrome de Bloch-Sulzberger (Incontinentia pigmenti). Características y aporte de un caso clínico. Arch Argent Pediatr. 2011;109(3):62-5.
11. Lee Y, Kim S, Kim K, Chang M. Incontinentia pigmenti in a newborn with NEMO mutation. J Korean Med Sci. 2011;26:308-11.
12. O'Doherty M, McCreery K, Green AJ, Tuwir I, Brosnahan D. Incontinentia pigmenti-ophthalmological observation of a series of cases and review of the literature. Br J Ophthalmol. 2011;95(1):11-6.
13. Minic S, Trpinac D, ObradovicM. Systematic review of central nervous system anomalies in incontinentia pigmenti. Orphanet J Rare Dis. 2013;8:25.
14. Llano-Rivas I, Soler-Sánchez T, Málaga-Diéguez I, Fernández-Toral J. Incontinentia pigmenti. Four patients with different clinical manifestations. An Pediatr (Barc). 2012;76:156-60.
15. Minic S, Trpinac D, Obradovic M. Incontinentia pigmenti diagnostic criteria. Update. Clin Genet. 2014;85:536-42.
16. Berg AT, Berkovic SF, Brodie MJ, Buchalder J, Cross H, Emde Boas WV, et al. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia. 2010;51:676-85.
17. Fisher RS, Acevedo C, Arzimanoglou A, Bogaz A, Cross H, Elger CE, et al. A practical clinical definition of epilepsy. Epilepsia. 2014;55(4):475-82.
18. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: Position paper of the. ILAE Commission for Classification and Terminology. Epilepsia. 2017;51(4):1-10.
Recibido: 24 de enero de 2017.
Aprobado: 7 de enero de 2018.
Ernesto Portuondo Barbarrosa. Hospital Pediátrico Centro Habana. Calle Benjumeda y Morales, municipio Cerro. La Habana, Cuba. Correo electrónico: ernestopb@infomed.sld.cu


{kind=link}
{kind=link}