










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El síndrome de Goodpasture (SGP) es una enfermedad autoinmune, inusual en pediatría, provocada por anticuerpos anti membrana basal glomerular (Ac-AMBG), de causa inmunológica, se caracteriza por la presencia de anticuerpos circulantes, que actúan directamente contra un antígeno intrínseco de la membrana basal glomerular y provoca una glomerulonefritis rápidamente progresiva (GNRP).1) Descrita desde 1919 por el Dr. Ernest Goodpasture como GNRP, asociada con hemorragia alveolar en un paciente de 18 años con diagnóstico de influenza. En 1958 se acuñó su nombre actual, síndrome de Goodpasture, para describir una enfermedad con afección renal, mediante el desarrollo de glomerulonefritis asociada con hemorragia de origen pulmonar.2,3 Las manifestaciones clínicas corresponden a una GNRP y a hemorragia alveolar que determinan la evolución de la enfermedad, pues corresponden a la principal causa de muerte de estos pacientes. Los Ac-AMBG (típicamente IgG, pero en algunas oportunidades IgA o IgM) están dirigidos contra el dominio NC1 de la cadena alfa-3 del colágeno tipo IV,4,5,6,7,8 Esta molécula se expresa en mayor medida y con mayor accesibilidad en la membrana basal del glomérulo renal y de los alvéolos pulmonares, pero también en los túbulos renales, plexos coroideos, capilares retinianos y la membrana de Bruch. Bajo condiciones normales, el endotelio alveolar (carente de fenestraciones) es una barrera contra los Ac-AMBG pero existen factores que aumentan su permeabilidad al ingreso de los anticuerpos; estos son: aumento de la presión hidrostática, mayor fracción inspirada de oxígeno, exposición a hidrocarburos, infecciones y el tabaquismo.1,6) La enfermedad se desencadena mediante la asociación de estos factores, con susceptibilidad genética del huésped (alelos HLA DRw15, HLA DRB*03, DRB*04).7 presentes en más del 80 % de los casos. La unión antígeno-anticuerpo activa la cascada del complemento, estimula el reclutamiento de células inflamatorias, la actividad de enzimas proteolíticas y produce daño en los órganos afectados.
El SGP es extremadamente raro, un caso por 1 000 000 de personas por año, es infrecuente en la infancia. Predomina en la segunda a tercera década y en la quinta a sexta década de la vida, con ligero predominio en varones.6) Consideramos un caso atípico, inusual en esta edad, se trata del primer caso pediátrico publicado en Cuba, lo que motivo su presentación.
PRESENTACIÓN DEL CASO
Preescolar femenina de 6 años, piel blanca, con antecedente de diez hospitalizaciones desde los cinco meses de edad, en las que predominaron los signos y síntomas de un síndrome obstructivo bronquial difuso bilateral, aumento del diámetro antero posterior torácico, tiraje bajo ligero, hipersonoridad y estertores secos con fiebre, sin elevación de reactantes de fase aguda y con radiografías de tórax interpretadas como procesos bronconeumónicos de predominio en vértice de pulmón derecho e hilio basal bilateral (Fig. 1), tratados con broncodilatadores, penicilina parenteral y esteroide oral. En la penúltima hospitalización se le diagnosticó además sepsis severa asociada a micosis respiratoria y se trató con antibióticos de amplio espectro y requirió adicionalmente transfusión sanguínea (Hb: 66 g/L).
La paciente fue ingresada en nuestra institución a los cuatro años, por presentar un cuadro clínico de quince días de evolución, caracterizado por hematuria macroscópica y “orina espumosa”, asociado a edema de miembros inferiores referidos por la familia, adicionalmente, palidez mucocutánea. Recibía tratamiento con fluconazol oral, que había sido prescrito en la hospitalización previa.
Se obtuvo el consentimiento informado por escrito de la paciente, para la publicación de este caso.
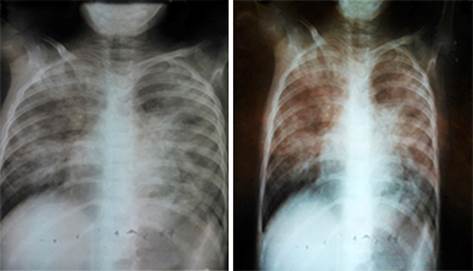
Fig. 1 Rayos x de tórax en proyección antero posterior. Muestra infiltración reticular intersticial que compromete vértice derecho y tercio medio de ambos pulmones.
Examen físico
La preescolar presentaba buenas condiciones generales, palidez mucocutánea generalizada, peso: 20 kg, talla: 115 cm (valoración nutricional: percentil 50-75, normopeso), frecuencia cardíaca de 90 latidos por minuto sin soplos auscultables y respiratoria de 22 respiraciones por minuto, tensión arterial 90/60 mmHg, con saturación arterial de 97 % por oximetría de pulso. Los hallazgos clínicos cardiorrespiratorios de la paciente mostraron una buena expansibilidad torácica con incremento del diámetro anteroposterior sin tiraje; hipersonoridad y a la auscultación murmullo vesicular conservado con estertores secos: roncos y sibilantes aislados. El examen físico abdominal fue normal, no se palparon masas ni visceromegalias. Tampoco se encontraron edemas de miembros inferiores. Durante su hospitalización, la paciente siempre presentó un ritmo diurético de 1,5 mL/kg/hora, con hematuria macroscópica de característica glomerular. El resto del examen físico no presentó alteraciones.
Complementarios
Los estudios realizados el primer día de estancia hospitalaria, mostraron un hemograma con leucocitos en 8,9 x 109/L; polimorfonucleares 64 %; linfocitos 27 %; monocitos
4 % y eosinófilos 5 %. hemoglobina: 43 g/L; reticulocitos de 34 x 10-3/L; plaquetas en 285 x 109/L; velocidad de sedimentación globular de 43 mm/h y una lámina periférica que reportó hipocromía +++/++++; microcitosis y algunos ovalocitos; VCM: 90,3 fl; HCM: 24,9 pg; CHCM: 276 g/L; glicemia: 5,4 mmol/L, alanintransaminasa: 5,3 U/L; aspartatotransaminasa: 19 U/L; colesterol total: 4,8 mmol/L, triglicéridos: 1,94 mmol/L, proteínas totales: 69 g/L y albúmina: 39,3 g/L.
Las pruebas de función renal arrojaron los siguientes resultados: creatinina: 53,3 µmol/L, proteinuria: 3,66 g/24h, relación proteína creatinina: 9,55g/g; densidad urinaria:1 018, filtrado glomerular: 95 mL/min/1,73 m2sc; cituria: 250 000 hematíes/mL; pH: 6 y urocultivo negativo.
Al cuarto día de estancia hospitalaria se realizó discusión conjunta del caso con las especialidades pediátricas de neumología, inmunología, hematología y nefrología y se decidió realizar los siguientes estudios diagnósticos:
Hisopado faríngeo profundo: Staphylococus aureus ++++; micológico y esputo BAAR: negativos; electrolitos en sudor: NaCl: 24 mmol/L; Mantoux: negativo; dosificación de inmunoglobulinas: IgA: 2,53 g/L, IgG: 9,47 g/L, IgM: 1,81 g/L y complemento sérico: C3: 1,08 g/L; C4: 0,33 g/L.
Imágenes
Ultrasonido abdominal y renal: riñón derecho de 86x38 mm, con aumento de la ecogenicidad del parénquima renal que midió 8 mm, sin dilatación del sistema excretor. Riñón izquierdo de características similares al riñón derecho con 84x40 mm, contornos lobulados con parénquima de 7 mm.
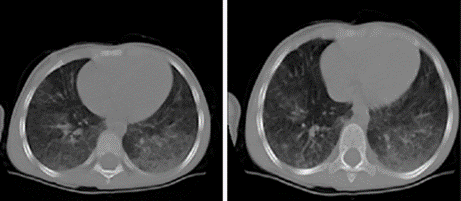
Tomografía computarizada simple de tórax: cortes axiales a 5 mm que permitió observar densidades difusas de aspecto granular fino de localización centrolobulillar con engrosamiento del espacio broncovascular que sugirió patrón en vidrio deslustrado, lo que puede corresponder con el grupo de neumopatías intersticiales difusas de localización basal posterior bilateral, no asociada a alteraciones pleurales (Fig. 2).
Estudios adicionales
Fibrobroncoscopia con lavado broncoalveolar y biopsia: estudio bacteriológico sin crecimiento bacteriano; biopsia: presencia de histiocitos abundantes cargados de hemosiderina y escasas células cargadas de grasa; coloraciones especiales positivas para hemosiderina y grasa
Adicionalmente, se decidió realizar estudios inmunológicos, con los siguientes resultados: anticuerpos anticitoplasma de los neutrófilos (ANCA) (cribado anti-PR3 y MPO): positivo; anti-PR3 (cANCA) método ELISA: 2 U/mL (valor de referencia: 0-5 U/mL); anti-MPO (pANCA) método ELISA: 14 U/L (valor de referencia: 0-5 U/mL); Ac-AMBG (ELISA): 53 U/mL (valor de referencia: 0-5 U/mL).
Evolución clínica
De acuerdo a los hallazgos clínicos y de laboratorio, al cuarto día de ingreso hospitalario se transfundió sangre fresca total (200 mL). Durante el décimo tercer día de estadía en sala, la paciente presentó tos seca paroxística no hemoptoica, asociada a febrícula y cefalea global. El examen físico reveló la presencia de palidez mucocutánea generalizada, congestión nasal, rinorrea acuosa, polipnea ligera, tiraje subcostal leve y sibilancias universales a la auscultación, con saturación de 98 % comprobada mediante la oximetría de pulso. El examen físico neurológico fue normal. Se indicó oxígenoterapia a 5 litros/minuto, aerosoles cada 6 horas con broncodilatador de acción rápida (salbutamol: 0,01 mg/kg/dosis) y se realizó hemoglobina cuyo resultado fue de 54 g/L, por lo cual se decidió transfundir nuevamente (200 mL de sangre fresca total).
Se inició tratamiento inmunosupresor después de los resultados del estudio inmunológico, en correspondencia con las manifestaciones clínicas de la niña. indicándosele un primer ciclo de inducción con esteroides por vía endovenosa (metilprednisolona 30 mg/kg/dosis, 3 dosis), asociados a ciclofosfamida: 500 mg/m2sc/día una vez al mes y continuar con esteroides por vía oral.
Los síntomas respiratorios desaparecieron, se egresa con tratamiento ambulatorio y seguimiento por la especialidad de nefrología. El tratamiento fue rápidamente efectivo con una disminución inmediata en los títulos de Ac-AMBG. Durante 2 años la niña se mantuvo libre de síntomas respiratorios, con cifras de hemoglobina normales, no hematuria, solo persistió proteinuria ligera, evolutivamente la proteinuria aumento llegando a un gramo en 24 horas, por lo que se consideró criterio para la realización de biopsia renal.
Reporte de la biopsia
Se obtuvo cilindro con 33 glomérulos, 8 de ellos con esclerosis total y parcial, en 5 de ellos atrofia tubular focal con engrosamiento de la membrana basal tubular e infiltrado inflamatorio focal ligero intersticial de células mononucleares. No se observó daño vascular.
Inmunofluorescencia: se observó depósitos de IgM + en zonas con esclerosis, IgA, IgG, C1q negativos.
Diagnóstico anatomopatológico: Glomeruloesclerosis segmentaria y focal.
DISCUSIÓN
El SGP es una enfermedad de base inmunológica, caracterizado por la tríada de GNRP, presencia de anticuerpos circulantes Ac AMBG y hemorragia pulmonar. Entre un 10 y un 30 % de los casos, la enfermedad por Ac AMBG se asocia con ANCA positivo y la mayoría de los pacientes tienen títulos bajos de anticuerpos antimieloperoxidasa (MPO). Este subgrupo de pacientes probablemente presente una variante de vasculitis asociada, sin embargo, aunque no se ha esclarecido cuál es la causa exacta del desarrollo de ANCA en la enfermedad por Ac-AMBG, algunos autores han sugerido que un mecanismo de activación policlonal podría ser el responsable.1 El diagnóstico diferencial del SGP incluye las diferentes formas de vasculitis sistémicas que pueden comprometer pulmón y riñón, el término síndrome pulmón-riñón describe la presencia de hemorragia alveolar difusa y glomerulonefritis como manifestación de algunas enfermedades autoinmunes multisistémicas entre ellas la granulomatosis de Wegener, el síndrome de Chug-Strauss, la poliarteritis nudosa, la esclerosis sistémica progresiva y el lupus eritematoso sistémico. Todas ellas son infrecuentes en la edad pediátrica, pero se pueden asociar a hemoptisis, la que raras veces es significativa.(5,8,9) Dentro del protocolo de diagnóstico se requieren niveles de ANCA, broncoscopia, tomografía de tórax y estudio de sedimento urinario.10) Las determinaciones en suero del nivel de complemento, del título de Ac-AMBG, anticuerpos antinucleares, crioinmunoglobulinas e inmunocomplejos circulantes, permiten el diagnóstico por métodos incruentos de las entidades citadas. Las tres causas más comunes de presentación del síndrome pulmón riñón son las vasculitis ANCA+ de vasos pequeños asociados a (Ac-AMBG), el SGP y el lupus eritematoso sistémico (LES).11
El diagnóstico definitivo se realiza mediante toma de biopsia, sin embargo, la existencia de Ac-AMBG mediante la prueba ELISA puede incluirse en el diagnóstico con buena sensibilidad.3) La biopsia renal permite identificar la lesión histológica común: la proliferación de las células epiteliales del espacio urinario del glomérulo con formación de las características semilunas de distribución difusa, acompañadas de lesiones de necrosis fibrinoide de distribución preferentemente focal y segmentaria. El estudio del material de biopsia por inmunofluorescencia permite aislar la enfermedad por anticuerpos antimembrana basal del resto de las entidades. La paciente que estudiamos reúne ciertas particularidades como el comienzo de la enfermedad en edad temprana de la vida, el inicio con manifestaciones respiratorias inespecíficas, caída brusca de la hemoglobina, hematuria y la ausencia de fallo renal agudo. Se presentó con la asociación de anemia severa, microcítica, hipocrómica, con infiltrados pulmonares bilaterales, de lo que surge el diagnóstico clínico-radiológico de hemosiderosis pulmonar. (12,13 Esto se confirmó por la presencia de histiocitos abundantes cargados de hemosiderina. Inicialmente se difiere biopsia renal por la afectación respiratoria que presentaba y se tuvo en cuenta además la elevada sensibilidad y especificidad de la presencia de Ac-AMBG para el diagnóstico de dicho síndrome, registrado en investigaciones consultadas.3
Justificada por la persistencia de la proteinuria, en aumento, se realiza biopsia renal, donde se confirma glomerulopatía crónica, en este caso glomérulo esclerosis segmentaria y focal. La ausencia de los depósitos típicos de anticuerpos a largo de la membrana basal, se pudiera explicar porque la biopsia se realizó, tras el tratamiento inmunosupresor, cuando los Ac- AMBG eran imperceptibles.
Podemos concluir que por tratarse de una entidad rara en pediatría se requiere un diagnóstico temprano y tratamiento agresivo para mejorar el pronóstico del paciente. En su seguimiento son necesarias una terapia farmacológica prolongada, una adecuada adherencia al tratamiento propuesto y un estrecho monitoreo clínico y analítico de lo cual dependerá la progresión de la injuria renal y pulmonar.

