










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La osteogénesis imperfecta (OI),1,2 o enfermedad de Lobstein o enfermedad de los huesos de cristal3 o huesos quebradizos, es la displasia esquelética más común, afección de causa genética, de baja prevalencia, considerada enfermedad rara.4 Comprende un grupo heterogéneo de síndromes del tejido conectivo, con el rasgo congénito de fragilidad ósea.5 La severidad de las manifestaciones clínicas es variable, desde forma leve hasta severa o letal en relación con los distintos tipos descritos. Por ser tan débiles los huesos se quiebran fácilmente, lo que ocurre sin poder reconocer una causa específica, o en ocasiones producido generalmente por golpes suaves o cualquier caída, por lo que las fracturas múltiples son frecuentes, y pueden suceder incluso antes del nacimiento.1,4,5,6
Los estudios en años recientes de genética molecular han confirmado la heterogeneidad de la OI,7 a través de más de 150 mutaciones de los genes que codifican la síntesis del colágeno tipo I (COL.1A1, cadena alfa I) en el cromosoma 17 y en el cromosoma 8 (del COL.1A2, cadena alfa II), de acuerdo a lo postulado por Sillence en 1979,8) quién estableció la clasificación inicial para los distintos tipos de la enfermedad fundamentada en su descripción clínica, 7,8 con herencia variable, y por otros genes descubiertos a posteriori.9,10,11
El colágeno tipo 1 es el más abundante del cuerpo y constituye parte importante de la estructura y función del corazón, los pulmones, el esqueleto, y otros órganos. Las personas con OI tienen menos colágeno de lo normal o un colágeno de calidad menor.(7,12 Aproximadamente el 85-90 % de los individuos son heterocigotos por la referida mutación en uno de los genes COL. 1A1 y, COL.1A2,4,9) sin embargo, un amplio rango de mutaciones recesivas (AR) esporádicas y más infrecuentes, debidas en especial a los genes CRTAP (codifica a la proteína asociada al cartílago, sigla en inglés), LEPRE1, SERPINF1, BMP1 y TMEM388 y IFITMS, entre otros descritos en años recientes, son determinantes de la OI.1,2,12,13,14,15,16
Está asociada la OI con debilidad muscular e hiperlaxitud de ligamentos y articulaciones (al flexionar o extender de manera exagerada las articulaciones con hipermovilidad) y deformaciones óseas.(2,16) Otros síntomas son las denominadas articulaciones sueltas y malformaciones esqueléticas, incluído afectación en la base del cráneo,17,18 signos de retraso del crecimiento, huesos y columna vertebral curvas,19,20,21,22 en algunos tipos escleróticas azules o grises23) y enfermedades cardiovasculares y pulmonares.24,25,26
Entre las manifestaciones extra-esqueléticas, en los dientes se produce dentinogénesis imperfecta, alteración autosómica dominante del desarrollo odontológico, lo que provoca dentadura decolorada, amarilla, por dentina pobre y opalescente, que se gasta con facilidad, caries frecuentes, con bajo contenido mineral y canal pulpar obliterado. 1,7 Otras manifestaciones son la pérdida de la audición /tipo I y III en edad adulta.5,6).
El objetivo de este trabajo es presentar un paciente con las características clínicas e imagenológicas de osteogénesis imperfecta de tipo III.
PRESENTACIÓN DEL CASO
Se trata de un niño de 4 años de edad, nacido en la República del Ecuador, residente en la localidad de Palama, perteneciente a la zona rural de Mulliquindi Santa Ana, de la cordillera de Los Andes ecuatoriana, provincia de Cotopaxi, con peso al nacer de 2 850 g, por parto eutócico, hijo de padres aparentemente sanos no consanguíneos, con historia de múltiples fracturas óseas espontáneas, no relacionadas con traumas, a partir de los 8 meses de edad, y dos meses después nueva fractura sin causa aparente. En el curso de su vida se registran 16 fracturas, en especial en huesos largos, costillas y vértebras; no antecedente familiar de osteogénesis imperfecta. Al examen físico niño orientado en tiempo y espacio, sociabilidad con su medio familiar normal, no deambula, baja talla, cara de forma triangular, retardo en erupción dentaria, ausencia de dentinogénesis: escleróticas de color normal, aunque la madre refiere la esclera presentó un tinte azul hasta los 8 meses de edad. Son rasgos físicos los brazos y piernas arqueadas, y deformidad torácica por columna vertebral con curva por cifosis y escoliosis (curvatura de la columna vertebral en forma de “S”) y esternón prominente.
Los estudios radiológicos de huesos largos y columna vertebral muestran los siguientes aspectos: huesos largos traslúcidos con ensanchamiento brusco en las epífisis y cortical delgada, fémur en su extremo distal y proximal de la tibia, con esclerosis. Hay pérdida del trabeculado óseo normal. En ambos fémures se aprecia deformidad marcada en las epífisis, en genus valgus con fracturas y áreas de esclerosis de la cortical. Además, se aprecia ensanchamiento de los huesos largos, con tibias y fémures con cortical delgada (afinamiento) y evidente pérdida de trabeculado óseo normal (Fig. 1 y 2).
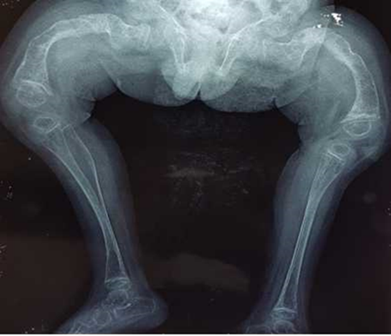
Fig. 1 - Ambos fémures con deformidad marcada en las epífisis, con fracturas y áreas de esclerosis. Ensanchamiento de los huesos largos, tibia y fémur con cortical delgada, hay pérdida del trabeculado óseo normal. (Edad 2 años y 11 meses, rayos X realizado en elCentro Médico Área de Salud de Latacunga, Ecuador).

Fig. 2 - Marcada deformidad de ambos fémures en genus valgus con pérdida del trabeculado óseo normal y con ensanchamiento de las epífisis, hay esclerosis de la cortical y deformidad en ambas epífisis. (Edad 2 años y 11 meses, rayos X realizado en Centro Médico Área de Salud.de Latacunga. Ecuador).
En la caja torácica hay marcada deformidad con prominencia del esternón y la columna dorso lumbar con escoliosis a doble curva, osteoporosis con adelgazamiento de la cortical y fracturas costales antiguas con áreas de esclerosis en las últimas costillas derecha (Fig. 3).
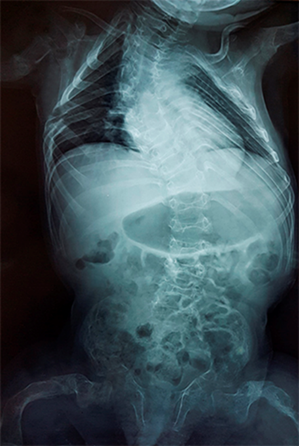
Fig. 3 - Marcada deformidad de la caja torácica y la columna dorso lumbar con escoliosis a doble curva, osteoporosis con adelgazamiento de la cortical y fracturas costales antiguas con áreas de esclerosis en las últimas costillas derecha. (Edad 3 años y 11 meses, rayos X realizado en Hospital General Pedro Arturo Suárez. Quito, Ecuador).
Las manifestaciones clínicas descritas han sido progresivas en el transcurso de la infancia. Al momento actual el niño recibe tratamiento con calcio y vitamina D 800 U (Calcibon D, 800 U), desde hace 8 meses. No ha sido sometido aún a tratamiento con bifosfonatos.
La sintomatología clínica y hallazgos radiológicos permiten afirmar el diagnóstico de OI, clasificado como tipo III y la evaluación del genetista clínico confirmó el diagnóstico.
DISCUSIÓN
La historia del niño con múltiples fracturas y deformidades óseas en los huesos largos, en especial ambos fémures, y en la columna vertebral con escoliosis severa en forma de “S” causada por múltiples fracturas debido a fragilidad ósea, son manifestaciones clínicas compatibles con el diagnóstico de OI, enfermedad de baja prevalencia o rara (ER), pues afecta pequeño número de personas, en comparación con la población general, y su propia rareza plantea cuestiones específicas.2,4,27) La Unión Europea describe que afectan a menos de cinco personas por diez mil habitantes y reconoce alrededor 7 000 ER.28) La OI es una enfermedad con el rasgo de ser grave, crónica y progresiva, con expresión desde la etapa prenatal, y manifestaciones clínicas al nacimiento,29 infancia30,31 y edad adulta.32
En 1979, Sillence8 al establecer la clasificación definió cuatro grupos basado en la descripción clínica y severidad de la enfermedad,6) casi exclusivamente sustentado en el fenotipo evaluado por el médico, con formas de herencia dominante (AD) o recesiva (AR). De inicio se clasificó en cuatro tipos: OI tipo I, leve, común, no deformante con esclera azul;33) OI tipo II, forma grave perinatal, letal en el primer año de vida,34) presenta además, insuficiencia cardíaca; OI tipo III severa, progresivamente deformante, con esclera de color normal;35) OI tipo IV severidad moderada con esclera normal. En 2009 se adicionaron los tipos V y VI. En 2014 la clasificación es actualizada hasta 11 tipos, y a posteriori a 15 tipos, basada en la herencia, fenotipo y genética. Se han descrito defectos en proteínas con funciones muy diferentes, que van desde el transporte estructural al enzimático, y desde el transporte intracelular a las chaperonas, por lo que es importante la identificación del gen afectado (tabla).9,31)
Tabla Clasificación osteogénesis imperfecta
| Tipos OI | Herencia | Fenotipo | Defecto genético |
|---|---|---|---|
| Tipos clásicos - | |||
| I | AD | Ligero | Nulo alelo COL.1A1 |
| II | AD | Letal | COL.1A1 o COL.1A2 |
| III | AD | Deformidad progresiva | COL.1A1 o COL.1A2 |
| IV | AD | Moderado | COL.1A1 o COL.1A2 |
| Causa desconocida | |||
| V | AD | Identificación histológica | IFITM5 |
| Defecto mineralización | |||
| VI | AD | Defecto mineralización Identificación histológica | SERPINF1 |
| Defecto 3-hidroxidación | |||
| VII | AR | Severo (hipomórfico): letal | CRTAP |
| VIII | AR | Severo o letal | LEPRE 1 |
| IX | AR | Moderado o letal | PPIB |
| Defecto chaperonas | |||
| X | AR | Severo o letal | SERPINH1 |
| XI | AR | Deformación progresiva (Síndrome Bruck 1) | FKBP10 |
AD: autosómica dominante; AR: autosómica recesiva. Fuente: Referencia bibliográfica 31.
El diagnóstico se ha categorizado y establecido la severidad sobre bases clínicas y radiológicas. El caso se clasifica como OI de tipo III fenotipo deformante de herencia dominante.31,35) Se descarta el tipo IV, pues las malformaciones óseas son más progresivas y deformantes en el tipo III. Las deformaciones óseas descritas, donde el grosor de la cortical está disminuido al igual que la cantidad de trabéculas óseas, y la baja talla, son indicadores de severidad. La evolución es invalidante, determinada por la aparición de fracturas desde la infancia con las consiguientes malformaciones óseas en el curso de la vida.36
Se describe OI en 1/10 000 a 1/ 20 000, y hasta 1/60 000 nacidos vivos.15) Puede afectar por igual a ambos sexos (proporción hombre mujer es 1:1) y sin diferencia de color de la piel; 35 % sin antecedentes familiares. En EE. UU., la prevalencia es 0,8 por 10 000 habitantes,37 en España la Asociación de Huesos de Cristal registra 0,6-1 por cada 10 000 habitantes,38) y en la comunidad Valenciana es inferior, con 0,29.15) En Dinamarca es 1,06 para similar proporción en población de habitantes.39 Es una enfermedad de distribución mundial, se ha descrito en las poblaciones de todos los continentes, en Suramérica se destacan informes de Argentina,40 Chile,33,34) Brasil,5) Venezuela;41). en Centroamérica Costa Rica42) y Cuba en el Caribe.43) En Asia, Taywan 44) y Viet Nam45 y en la población indígena de África negra de Sudáfrica,46) merecen destacarse. En Ecuador se estima en 1 caso cada 12 000-15 000 nacimientos, por tanto, si hay 350 000 nacimientos anuales, nacerán cada año aproximadamente 30 niños con OI y podría haber 1 166 casos nacionalmente, donde el alto grado de consanguinidad de su población, puede significar factor de riesgo, según publica la Guía Clínica de Diagnóstico y Tratamiento de OI del Ministerio de Salud Pública del Ecuador.47
La evolución en el niño ha sido invalidante, como sucede en las formas más graves, con afectación para toda la vida, al no deambular, y depender de una silla de rueda, por la condición de afectación incurable.1,4,5) La administración de tratamiento endovenoso con bifosfonatos ha abierto una nueva perspectiva terapéutica,48,49,50) aunque aún el caso presentado no ha recibido dicha terapia. Estos son compuestos sintéticos análogos de los pirofosfatos inorgánico con efecto de aumento de la densidad ósea y disminución del número de fracturas.51) En relación con el pronóstico, aquellos que no logran en la infancia alcanzar una adecuada masa ósea, en la edad adulta continúan padeciendo de fracturas, y posteriormente etapas de inmovilización permanente.
Es importante resaltar que, en la actualidad en el ámbito internacional, se han creado fundaciones y organizaciones de familias y grupos de enfermos en distintas latitudes que concentran a estos pacientes con el objetivo de brindar una mejor calidad de vida por ser más vulnerables en el ámbito psicológico y social y para apoyo terapéutico por el costo económico de la OI.39 En el país existe la Fundación Ecuatoriana de Osteogénesis Imperfecta (FEOI), organización que apadrina con sus acciones a más de 80 niños de familias de escasos recursos económicos, como es el caso de esta presentación, para garantizar su atención especializada.52
Se enfatiza el desafío de la calidad de vida de los niños con formas severas de OI. El enfrentamiento al niño con fracturas múltiples requiere de acciones específicas en distintas direcciones con el objetivo primordial de mejorar su capacidad física, lograr su movilidad e incorporación a la vida social.53) Los cambios establecidos en el manejo de OI basado en estrategias terapéuticas (fisoterapia, cirugía ortopédica y fármacos), siguiendo atención individualizada y relacionando los cuidados al grado de afectación del paciente representan posibilidades objetivas para la atención de la enfermedad.
Se realizó descripción clínica y radiológica de osteogénesis imperfecta, afección poco conocida, correspondiente al fenotipo III de la enfermedad, reportada en niño ecuatoriano de 4 años de edad, con talla baja que no deambula, expresión de la severidad de su afección genética, con severas alteraciones óseas por su fragilidad con fracturas múltiples en huesos largos y vértebras. Se llama la atención sobre la importancia del diagnóstico precoz, para establecer tratamiento específico y medidas de atención y control de la enfermedad.

