










 Servicios personalizados
Servicios personalizados Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Neurodevelopmental disorders (NDD) are featured by a delay in the acquisition of motor functions, cognitive abilities and speech, or combined deficits in these areas with the onset before the age of 5 years. The prevalence may reach 1-3 %, according to different studies1,2. Genetic causes account for approximately a half of all NDD cases; chromosomal aberrations (CA) account for 5-10%of cases3,4. However, the extensive variability of causal genetic defects hinders the etiological diagnosis. The phenotypic effect of CA depends on the type, origin, and length of aberrations. Additionally, gene density and functional aspects of genes and their interactions with other genes and environmental modulating significantly contributes to the phenotype, as well.5,6The aim of this work is to describe some alterations of the genome implied in neurodevelopmental disorders.
Methods
Bibliographic search in Medline, Pubmed, Scielo, LILACS and Cochrane, emphasizing in the last five years, the relationship between the various genetic factors that may be involved in neurodevelopmental disorders.
Results
Multiple genetic factors are involved in neurodevelopmental disorders, from gross ones such as chromosomal aneuploidies to subtler ones such as variations in the number of copies in the genome. Special emphasis is placed on microdeletion-micro duplication syndromes as a relatively frequent cause of NDDs and their probable mechanisms of formation are explained.
Microdeletion and microduplication syndromes
During the last decades an increasing number of so-called contiguous gene syndromes (CGSs) have been identified in patients with intellectual disability. CGSs are caused by an aberrant copy number (gain or loss) of specific chromosomal regions. Originally, CGSs were considered to encompass critical regions of two or more genes, located in a close proximity to each other. Some of these regions affect many genes but a limited number of genes may be dosage-sensitive and, thereby, causative for specific manifestations. Thus, the denomination “CGS” has been replaced by the designation “microdeletion or microduplication syndromes” (MMSs).7) The common molecular cause of recurrent MMSs is a non-allelic homologous recombination. Repetitive elements are prone to illegitimate intra- or interchromosomal/chromatide recombination during meiosis and mitosis. The expected number of MMS is suggested to be higher than is currently known, because many of these events have a deleterious intrauterine effect.8,9
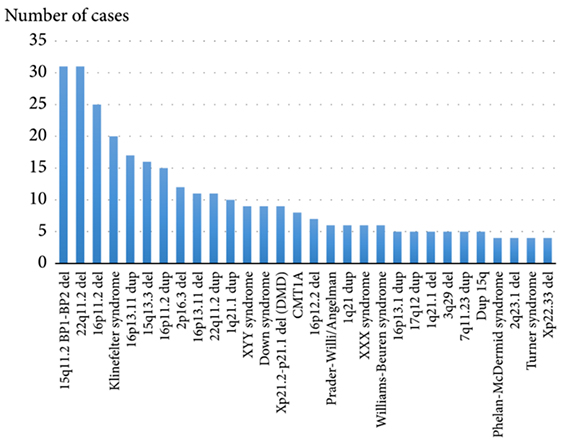
Theoretically, there should be a reciprocal microduplication syndrome for each microdeletion syndrome. However, microdeletion syndromes are more common according to current molecular cytogenetic studies (Fig.1). The main reason for this inequality is that microduplications are supposed to have a lower clinical impact than microdeletions. Subsequently, the patients with microduplications are less commonly addressed by molecular (cyto)genetic techniques due to less severe phenotypic manifestations.9,10
Additional causes for MMSs recurrence
Human non-homologous DNA recombination and end joining may occur due to double stranded breaks, which serve as a repair mechanism. This cause is less frequent and is not directly mediated by the features of the genome sequence.11In addition, MMSs can be a result of meiotic recombination when one parent carries a polymorphic inversion (e.g., a heterochromatic region of chromosome 9), which leads to a recombinant gamete with a deletion or duplication in this regiondue aninverted loop formation.9
Chromosomal aneuploidies
The most frequent cause of chromosomal syndromes is aneuploidy, which affects no less than 0.5% of newborns.12,13 Almost all of these conditions are associated with brain dysfunction 14 The most frequent aneuploidies among newborns are trisomy of chromosomes 21, 18, 13 and aneuploidy of sex chromosomes (gonosomes). Mosaic trisomy of chromosomes 8 and 9 (there are two cell populations (normal/trisomic) in an affected individual) are relatively common, as well (Tabla). 14,15

Copy number variations
Numerous human genome loci are prone to submicroscopic copy number variations (CNVs). These CNVs are located in euchromatic regions and dispersed over the entire human genome. CNVs(gain/loss of submicroscopic regions) can either lead to clinical manifestations or lack detectable phenotypic consequences.7,16 Several features of CNVs support their role in disease pathogenesis. Although less abundant than SNPs, they account for a larger nucleotide variation than SNPs because encompassing larger DNA sequences in the absolute size.17 Spanning thousands of nucleotide bases, CNVs often encompass and disrupt functional DNA sequences. Moreover, there appears to be enrichment in CNVs affecting “environmental sensor” genes, i.e. genes that are not necessarily critical for early embryonic development, but are involved in perceiving and interacting with the environment.18,19 he way how CNVs change their size when passed from one generation to the next, i.e. how specific DNA-regions are lost or amplified within a CNV, is not as yet completely understood. This may result from unequal crossing-over events in low copy repeat regions (5, 16), or from altered reparation after DNA-break and/or replication stress (e.g. non-allelic homologous recombination, microhomology-mediated break-induced replication, or chromothripsis).7,20,21,22,23,24 The most known disease-causing CNVs are 17q21.31microdeletions (Koolen-De Vries syndrome.25 reciprocal 17q21.31 microduplications,26,27 17q23.1q23.2 microdeletions28 and 3q13 microdeletions.29 Some microdeletion and microduplication syndromes are not caused by recurrent CNVs, but individuals witht he aberrations present with similar phenotypes,30 which allows for the definition of smallest regions of overlap, or critical regions, for the syndrome, which are likely to contain disease-causing genes (e.g. 1q43q44, 1q41q42 and 1q24q25 microdeletions).31,32,33
Genetic counseling
Genetic counseling in cases of CNVs with reduced penetrance and variable expressivity can be particularly difficult.34 If a parent carries the CNV, there is a 50% -inheritance-chance for off spring, but the establishment of additional risk factors in the family as well as the likelihood of having another affected childr equires more sophisticated (bioinformatics) analysis. For at least some recurrent CNVs, there are estimates either for penetrance, for any clinical phenotype or for psychiatric disorders, such as schizophrenia.35-36 The penetrance analysis shows that not all CNVs confer equal risk. steimates for the requency of carriers with significantly abnormal phenotypes range from ~10% for 15q11.2 BP1-BP2 microdeletion syndrome to~60% for distal 16p11.2 microdeletions.35) Unfortunately, it remains difficult to predict the severity of phenotypic outcomes, although some researchers suggest that there may be protective actors (e.g. gender)37.Furthermore, it is important to note that “penetrance” is quite relative; there may still be negative CNV effects among the so-called healthy parents or controls. This is supported by recent studies of general populations reporting negative effects of the CNVs, including cognitive impairments, other neuropsychiatric features, alterations in body weight, and reduced fertility.38,39 Therefore, it is likely that most of these CNV shave some type of negative impact on an individual’s development and neurologic functioning, although these impacts may not be always evident. Another notable feature of recurrent CNVs is variable expressivity. Same CNVs may be identified in populations of patients with intellectual disability, autism, and schizophrenia. Such effects may indicate that there are shared etiologies among NDD.40,41,42,43 Several studies have demonstrated the applicability of this assumption by comparing parental neurocognitive function to their children with de novo risk factor CNVs, showing a correlation between lower levels of parental functioning in specific domains (such as IQ and social responsiveness) scores and more significant impairments in those domains among their off spring.44,45.46
Final considerations
Genetic aberrations are found in at least 30-50% of children with NDD. Conventional karyotyping allows the detection of chromosomal aberrations en compassing more than 5-7 Mb, which represent 5-10% of causative genome rearrangements in NDD. Molecular karyotyping (e.g. SNP array/array CGH) can significantly improve the yield in patients with NDD and congenital malformations, which is repeatedly shown by systematic cohort studies. Recommendation: Cuban NDD cohorts have not yet been studied by these high-resolution techniques. The application of molecular karyotyping to the cohort can help in revealing new genetic aberrations and can provide effective genetic counseling.

