










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de Cushing es una afección endocrina de baja incidencia que resulta del exceso prolongado de cortisol.1,2 Puede presentarse a cualquier edad, no obstante, es poco frecuente en niños.2,3 La incidencia del síndrome de Cushing es 0,7-3,0 por cada 1 000 000 habitantes/año, de estos solo 10 % se presentan durante la infancia y la adolescencia.3,4,5,6,7 En EE. UU. la incidencia es relativamente alta, 49 por cada 1 000 000 habitantes/año.5
En niños tiende a presentarse con síntomas clínicos y la forma más común es la virilización en 55 % de los casos; el síndrome de Cushing sin virilización es poco frecuente (5 %).8 En estos pacientes se puede observar además un retraso del crecimiento. Tanto en adultos como niños la causa más frecuente del síndrome de Cushing es un tumor hipofisario productor de corticotropina (ACTH).2,3
Aproximadamente 75-90 % de los casos de síndrome de Cushing en niños se debe a la enfermedad de Cushing (de causa hipofisaria).2,3 La enfermedad de Cushing es poco común en niños menores de 6 años; las causas adrenales (adenomas, carcinoma o hiperplasia bilateral) son las etiologías típicas en niños.3
El carcinoma adrenocortical primario es muy raro y su incidencia anual varía entre 0,7-2,0 por cada 1 000 000 habitantes/año y es más frecuente su presentación en la cuarta y quinta década de la vida o edades muy tempranas.9,10,11 En su mayoría secretan solo glucocorticoides o glucocorticoides y andrógenos, siendo el carcinoma adrenocortical una causa rara de síndrome de Cushing. En los niños y adolescentes la incidencia es de 0,2-0,3 por cada 1 000 000 habitantes/año.9,10,11
Un estudio que evaluó la epidemiología de los tumores adrenales durante 23 años, determinó que el carcinoma adrenocortical solo representó el 0,3 % de la incidencia total y se presentó en uno de cada siete niños (de solo 13 infantes con tumores adrenales incluidos en el estudio).11
En Cuba no se han informado datos sobre la incidencia y mortalidad por tumores suprarrenales o síndrome de Cushing en niños. Recientemente se tuvo la experiencia de diagnosticar y tratar un caso relacionado con ambas afecciones que llegó al servicio de Endocrinología del Hospital Provincial General Docente “Dr. Antonio Luaces Iraola” de Ciego de Ávila, centro de atención secundaria de salud.
El objetivo de este artículo es describir los procedimientos diagnósticos y terapéuticos seguidos en el caso de una lactante de cinco meses de edad con síndrome de Cushing como consecuencia de un carcinoma adrenocortical.
Presentación del caso
Lactante femenina con cinco meses de nacida, procedente de un área rural de la provincia Ciego de Ávila (Cuba), hija de madre adolescente, producto de un parto eutócico a las 40,1 semanas de gestación, con peso al nacer de 3 400 g, sin enfermedad o ingreso previo y sin antecedentes patológicos familiares.
La madre de la bebé refirió notar un aumento excesivo de peso a partir del segundo mes de nacida, a pesar de presentar cierto grado de intolerancia a los alimentos, además de un pobre aumento en la talla, lo que motivó su consulta con el servicio de endocrinología y el equipo de endocrinología pediátrica.
En el examen físico se constató obesidad centrípeta y facie de “luna llena”. Presentaba un peso de 9,0 kg, talla de 57 cm y circunferencia cefálica de 44 cm. En la valoración del crecimiento se determinó un peso para la edad >97 percentil, peso para la talla >97 percentil y talla para la edad <3 percentil (según las Tablas Cubanas de Crecimiento y Desarrollo).
El cálculo de los valores del puntaje z según los estándares de la Organización Mundial de la Salud, arroja un peso para la talla con un valor de z= 6,42 DE (>+3DE), peso para la edad z=2,06 (>+2DE), talla para la edad z=-3,31 (<-3DE) y un IMC con valor z=5,78 (>+3DE). Todos estos valores fueron indicativos de mal estado nutricional de la paciente, que presentaba una ganancia excesiva de peso con pobre aumento de la talla.
En la piel se encontró atrofia cutánea con escoriaciones en ambos glúteos, ligero eritema facial y livedo reticularis. En el aparato respiratorio se constata murmullo vesicular audible, no estertores, con frecuencia respiratoria de 32/minuto. En el examen cardiovascular sus ruidos cardíacos eran rítmicos, de buen tono, pulso fuerte, buen llene capilar, con frecuencia cardíaca 130/minuto y tensión arterial 130/80 mmHg (>95 percentil). El abdomen se encontraba globuloso, blando, depresible y no impresionaba doloroso ni se palparon tumoraciones.
El examen del sistema nervioso mostró un cierre prematuro de fontanela anterior. Se observó agenesia de labios menores, sin vello axilar ni pubiano. Las características físicas de la paciente se muestran en la figura 1. La fotografía se tomó y se publica con el consentimiento de los padres de la lactante.
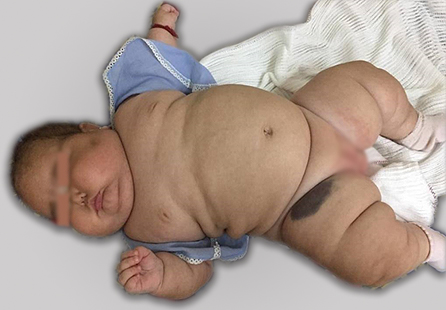
Fig. 1 Lactante de cinco meses con signos de hipercortisolismo endógeno. Obsérvese la obesidad y la facie pletórica (de luna llena) características.
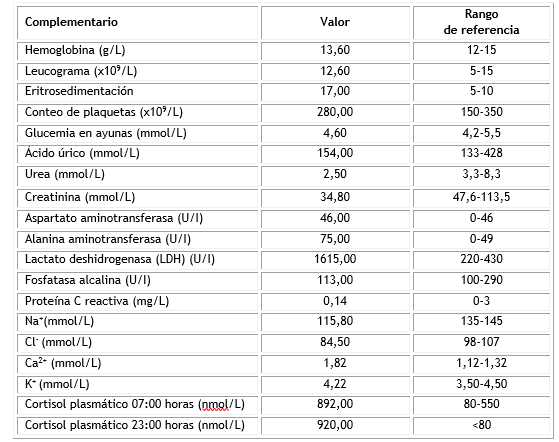
Ante la sospecha clínica de hipercortisolismo endógeno se indican los complementarios que se muestran en la tabla, donde se observan cifras elevadas de cortisol por encima de los valores de referencia, a las siete y 23 horas. También se encontró una elevación marcada de lactato deshidrogenasa y alteraciones hidroelectrolíticas.
Tabla Resultados de los análisis de laboratorio bioquímico y de función suprarrenal en el momento del diagnóstico
Por las características clínicas de la lactante y los valores de cortisol (ritmo de secreción de cortisol roto) se llega a la conclusión que se trata de un síndrome de Cushing, por lo que se procede a determinar la causa probable, para lo cual se indican varios estudios de imagen. Otros estudios hormonales y bioquímicos de utilidad para determinar la causa no estuvieron disponibles.
En el ultrasonido abdominal se aprecia un riñón derecho desplazado hacia abajo, de tamaño normal y parénquima sin alteraciones, en polo superior en proyección a la suprarrenal derecha se observa masa compleja predominantemente ecogénica que mide 52 x 42 mm, con zonas de necrosis y calcificación en su interior.
Se realiza tomografía axial computarizada (TAC) sin contraste, donde se observa un hígado homogéneo que rebasa 4,5 cm el reborde costal. En la proyección hacia la glándula suprarrenal derecha se observa imagen de aspecto tumoral con densidad variable entre 30 y 60 HU, de bordes bien delimitados que mide 65 x 47,5 x 40 mm, con desplazamiento de estructuras vecinas. No se encontraron adenopatías (Fig. 2).
Se le realiza además resonancia magnética nuclear de región selar, con la finalidad de descartar la causa hipofisaria del hipercortisolismo y ante la no disponibilidad de determinación de ACTH. Este procedimiento no reportó alteraciones.
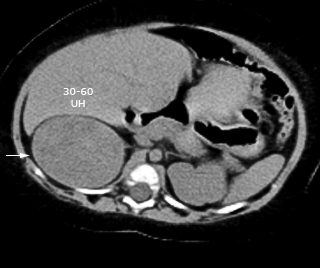
Fig. 2 Cortes axiales en tomografía axial computarizada, donde se observa imagen de aspecto tumoral (flecha) en proyección hacia glándula suprarrenal derecha.
Teniendo en cuenta la edad de presentación, el diámetro y la densidad mayor a 20 UH en las pruebas de imagen, se estableció como causa probable del síndrome de Cushing un tumor suprarrenal maligno.
La paciente es trasladada a un centro especializado donde se le realiza adrenalectomía unilateral derecha tomando muestra de la glándula para biopsia posoperatoria. Posterior a la cirugía, se indica tratamiento sustitutivo con esteroides (hidrocortisona) por insuficiencia suprarrenal, característica tras el proceder quirúrgico.
La biopsia posoperatoria (Identificador: CR2020-686) informa invasión capsular y vascular, necrosis extensa y mitosis atípicas, con conteo mitótico igual a 18 x 20 HPF, con inmunohistoquímica que determina CK positivo, melan A positivo, CD56 positivo, EMA negativo, Ki67 positivo en el 50 % de los núcleos tumorales y cromogranina negativo (Fig. 3). Estos resultados son compatibles, según la propuesta de Wieneke (12) para tumores pediátricos, con el diagnóstico de carcinoma adrenocortical. Según la clasificación de Sandrini13 la paciente se encontraba en el estadio 2 del carcinoma adrenocortical.
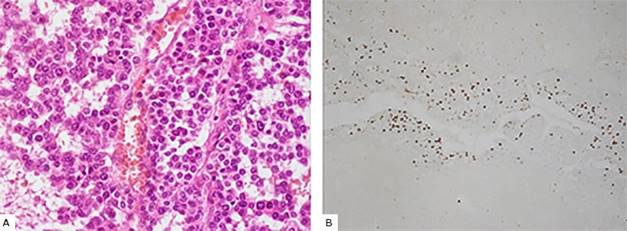
Fig. 3 Características histológicas del carcinoma adrenocortical. A. (Coloración con hematoxilina y eosina). Se observan las células tumorales con abundantes citoplasmas eosinófilos y grandes núcleos hipercromáticos con nucléolos prominentes; se presentan numerosas figuras mitóticas. B. Índice Ki67 con alto porcentaje de manchas nucleares (color marrón).
Pasado un mes de la cirugía, clínicamente se observa ligera mejoría, disminuye la obesidad y la hipertensión arterial remite, se sigue por consulta externa con tratamiento con hidrocortisona a dosis sustitutiva. No obstante, la lactante continua con intolerancia a los alimentos al punto de no poderle incluir el esquema de ablactación normal y solamente mantener la leche materna. Al final de ese mes la paciente es ingresada en la unidad de cuidados intensivos pediátricos ante sospecha clínica de insuficiencia suprarrenal. Se comprueba en el interrogatorio a familiares que existió un agotamiento del medicamento en los días previos al ingreso y al ser de un área rural se dificultó el acceso inmediato a nueva dosis, lo que precipitó el estado de insuficiencia suprarrenal aguda.
La lactante se presentó con deshidratación severa, difícil acceso venoso, deterioro marcado del estado general y cifras de cortisol plasmático más elevado que al momento del diagnóstico (950,00 nmol/L a las 07:00 horas), lo que indicaba metástasis tumoral. Padecía además un sangramiento digestivo por úlceras gástricas producidas por el exceso de cortisol. Finalmente fallece a los diez días de estadía hospitalaria como consecuencia de un fallo multiorgánico. En la necropsia se confirma presencia de células tumorales en vasos sanguíneos renales y pulmonares y en la grasa perirrenal derecha.
Discusión
El carcinoma adrenocortical en niños es un serio problema de salud con un pobre pronóstico si el diagnóstico se hace tardíamente. Su presentación en la infancia aumenta el riesgo de malignidad.14 En niños es frecuente que los tumores adrenales se presenten con excesos hormonales, siendo más común el aldosteronismo primario y el exceso de andrógenos que el síndrome de Cushing.13 La frecuencia de carcinoma adrenocortical y síndrome de Cushing en menores de un año es baja.2,9,10,11
En los casos de síndrome de Cushing en edad pediátrica, casi siempre se observa una detención del crecimiento. La talla suele estar por debajo de los valores normales y en cambio el IMC excede la media, tal como el caso presentado. Si por el contrario la talla y el peso aumentan a lo largo de la misma línea del percentil, resulta muy poco probable el diagnóstico de síndrome de Cushing. Otro aspecto indicativo de la enfermedad es la obesidad, que tiende a ser generalizada o centrípeta tal como se muestra en la figura 1.
La determinación plasmática de ACTH o cortisol en una muestra matutina aislada no es fiable, puesto que en sujetos normales estas hormonas se secretan de forma periódica según un ritmo circadiano que puede estar influido por el estrés físico o emocional. Los pacientes con síndrome de Cushing pueden presentar valores aislados dentro del intervalo normal, aunque la ausencia del nadir del ritmo circadiano condiciona un hipercortisolismo tisular.
El hipercortisolismo patológico se caracteriza por la pérdida del ritmo circadiano normal de la secreción de cortisol. Para demostrar esta alteración en la lactante se realizaron dos determinaciones de cortisol sérico, una de ellas en la mañana (07:00 horas) y la segunda por la noche (23:00 horas). No se hizo la prueba de supresión con dexametasona (1 mg).
En nuestra institución no contamos con la determinación de esteroides urinarios u otras mediciones hormonales indicadas en el estudio de estos casos, por lo que se confirmó el diagnóstico con los datos clínicos y las cifras de cortisol señaladas en la tabla 1.
Es importante medir andrógenos suprarrenales, teniendo en cuenta que es infrecuente que el carcinoma adrenocortical en niños tenga producción aislada de glucocorticoides, pues también produce andrógenos. Se consideró que no se trataba de un caso de hiperandrogenismo concomitante, pues a pesar de no disponer de estas determinaciones hormonales en el centro al momento del diagnóstico, el cuadro clínico no era compatible con un exceso de andrógenos suprarrenales.
Otras pruebas utilizadas para el diagnóstico del hipercortisolismo incluyen las siguientes:15
Cortisol libre en orina de 24 horas: se admiten como valores normales en niños los inferiores a 70 µg/m2/24 horas.
Cortisol plasmático a medianoche: en los niños, un valor de cortisol a medianoche superior a 121 nmol/L (4,4 μg/dl) identifica a los pacientes con síndrome de Cushing con una sensibilidad de 99 % y una especificidad del 100 %.
Cortisol salivar a medianoche: niveles de cortisol superiores a 27,6 nmol/L (1 μg/dl) al acostarse y a 7,5 nmol/L (0,27 μg/dl) a medianoche, confirman el diagnóstico.
Prueba de supresión con dexametasona 1 mg por la noche (prueba de Nugent): Esta prueba no ha sido estandarizada en niños y su principal inconveniente es que presenta 15-20 % de falsos positivos.
Existen otras pruebas dinámicas, como la prueba de supresión con dexametasona a dosis altas, la prueba de estímulo con hormona hipotalámica estimuladora de ACTH (CRH), prueba de estímulo con metopirona y de estímulo con vasopresina y sus análogos. No obstante, su disponibilidad es limitada en la atención médica fuera de centros de referencia.
Es importante señalar que no se debe efectuar ningún estudio radiológico hasta que se haya confirmado el síndrome de Cushing mediante el análisis hormonal. El punto de partida para estudiar a un paciente con una sospecha clínica seria de síndrome de Cushing son las características que mejor discriminan este trastorno. Una vez confirmado este síndrome se precisó la etiología con las pruebas de imagen (TAC) y posteriormente se confirma el diagnóstico de carcinoma adrenocortical en la biopsia pos cirugía. Otros marcadores biológicos no estuvieron disponibles para establecer el diagnóstico de carcinoma adrenocortical previo a la cirugía.
Según Wieneke, citado por Chatterjee y otros,12) para tumores pediátricos el diagnóstico de carcinoma adrenocortical se establece por los siguientes criterios:
Peso >400 g
Tamaño >10,5 cm
Invasión de vena cava
Invasión capsular
Invasión vascular
Extensión a tejidos blandos peri adrenales u órganos adyacentes
Necrosis
Índice mitótico >10 mitosis por 10 campos de gran aumento
Presencia de mitosis atípicas
Según estos criterios se considera tumor benigno cuando hay ≤2 criterios, tumor de comportamiento indeterminado cuando hay 3 criterios, y mal pronóstico si hay ≥4 criterios. La lactante del caso descrito presentaba cuatro criterios (mal pronóstico).
La edad menor de 18 años, el tamaño del tumor, la bilateralidad, el modo de descubrimiento y la densidad en unidades Hounsfield (≥20 HU) han sido asociados con un mayor riesgo de malignidad en el hallazgo de las masas adrenales.16 En el caso de nuestra lactante los factores asociados a malignidad presentes fueron la edad, el tamaño del tumor y la densidad en las pruebas de imagen (30-60 HU).
Para determinar el estadio del carcinoma adrenocortical se utiliza la clasificación establecida por Sandrini,13 y modificada por el Grupo de Oncología Pediátrica (COG, por sus siglas en inglés)17 basada en los datos procedentes del Registro Internacional de Tumores Adrenocorticales Pediátricos (IPACTR, por sus siglas en inglés), según el cual se clasifican en:
Estadio 1: tumor completamente resecado, con peso <100 g, volumen <200 cm³ y concentraciones hormonales posoperatorios normales.
Estadio 2: tumor completamente resecado, con peso >100 g, volumen >200 cm³ y concentraciones hormonales posoperatorios normales.
Estadio 3: tumor residual o inoperable.
Estadio 4: presencia de metástasis.
El caso de la lactante que se presenta, se encontraba en el estadio 2 según esta clasificación.
El tiempo medio de diagnóstico del carcinoma adrenocortical es de 5 meses (0-90 meses), a predominio del sexo femenino y antes de los 4 años,8 lo que coincide con este caso. Por otro lado, en el 90 % de los casos se presenta como asintomático, la forma más común es la virilización (55 %) y en menor frecuencia el síndrome de Cushing (5 %), siendo rara la presentación del síndrome de Cushing aislado.8)
La resección completa es la terapia de elección para el control de la enfermedad, tal como se realizó con la paciente, a pesar de lo cual fallece en el plazo de un mes pos cirugía. La mortalidad por carcinoma adrenocortical a los cinco años es de aproximadamente 75-90 %, teniendo en cuenta que el diagnóstico se realiza frecuentemente en un estadio avanzado y las opciones terapéuticas son desafortunadamente limitadas.18 Un estudio que incluyó los datos procedentes de la Base de Datos Nacional de Cáncer de los Estados Unidos (NCDB, por sus siglas en inglés) entre 1998 y 2011, encontró que los pacientes con carcinoma adrenocortical en edades pediátricas tenían peor supervivencia cuando la edad es ≥4 años, extensión de la lesión primaria a estructuras adyacentes y la presencia de metástasis.19 En el caso presentado, no estaban presentes ninguno de estos criterios en el momento del diagnóstico. Otros biomarcadores predictores de mal pronóstico18 no estuvieron disponibles en las instituciones de salud donde se ofreció asistencia médica a la lactante.
La metástasis tumoral del carcinoma adrenocortical se presenta con mayor frecuencia en niños mayores de 12 años en comparación con los menores de 4 años y son el hígado y pulmones los sitios más frecuentes,14 lo que no se corresponde con lo descrito en este caso a excepción de la metástasis en tejido pulmonar hallada durante la necropsia.
Es importante sospechar el carcinoma adrenocortical ante un niño con síndrome de Cushing o cualquier manifestación de hiperfuncionamiento adrenal o hipercortisolismo endógeno, de manera que el diagnóstico se haga precozmente, lo que reduce el tiempo entre el diagnóstico y el tratamiento quirúrgico. En este caso, a pesar de contar con equipos de atención pediátrica especializados, es poca la experiencia en la atención de tumores suprarrenales en la infancia, en parte por su baja incidencia.
El síndrome de Cushing en la edad pediátrica es una enfermedad poco frecuente, de etiología variada y sintomática. Cuando aparece la causa más probable es la suprarrenal y dentro de esta el carcinoma, si bien la presentación de estas entidades es rara. Este caso pone de manifiesto la importancia de la atención especializada y cuidadosa, así como la necesidad del diagnóstico precoz en hospitales de mediana complejidad, teniendo en cuenta que estos pacientes tienen un mal pronóstico. Se hace necesario conocer más sobre estos casos y tener en cuenta sus formas de presentación por infrecuente que puedan ser, basados en el precepto “lo que no se piensa, no se diagnostica”.
El caso presentado es de los pocos casos conocidos y primero publicado en Cuba de un síndrome de Cushing por carcinoma adrenocortical en edades pediátricas.

