










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El término enfermedades raras (ER) o poco comunes, surgió en la década de los años 80 del pasado siglo xx en EE. UU., y años después continuaba la expansión del concepto en Europa y otros países del orbe. Las ER se asociaban con la necesidad de nuevos tratamientos y medicamentos para enfermedades de baja prevalencia, evento decisivo en el surgimiento de la denominación de las ER. Estos medicamentos se catalogaban huérfanos, debido a que no resultaban rentables por su indicación limitada.1
Si bien no existía un acuerdo sobre la definición, se aceptaba que las ER constituyen aquellas de muy baja prevalencia, entre 6 y 8 %, en la población general.
Esta definición creaba contradicciones en la cuantificación de los casos. En EE. UU., se apoyaba en la frecuencia y la prevalencia, mientras que, en otros países de Europa, se consideraban otros factores como la gravedad del paciente y la calidad de vida.1) Estas contradicciones existieron con independencia de los diferentes umbrales de las enfermedades en distintos territorios y sus variaciones por países.2
Una enfermedad se consideraba rara en una región y de mayor frecuencia en otra, lo cual se debía, entre otros, a su origen genético, y, en ocasiones, se correspondía con la falta de pesquisa determinada por un criterio falso, y hasta por limitado conocimiento acerca de las formas de presentación y manifestaciones clínicas de la enfermedad.3
El 50 % de estas enfermedades se diagnosticaban en la infancia y la adolescencia, la mayoría de ellas aparecían durante los dos primeros años de vida y se les atribuía un porcentaje elevado de discapacidad y mortalidad infantil.1
La OMS reporta que el 7 % de la población mundial padece una enfermedad rara, que comprende globalmente a alrededor de 300 millones de personas entre niños y adultos: 25 millones en EE. UU., 30 millones en la población europea y 3 millones en España.4,5 En Ecuador, se estima un aproximado de 1 millón de personas afectadas con alguna de las enfermedades reconocidas raras a nivel mundial.6) Varios países de la región latinoamericana no llevan un control oficial de estas enfermedades.
En la actualidad, la base de datos Orphanet de la Unión Europea,7) documenta 6172 enfermedades como raras,2 de las cuales 79 % resulta de origen genético, con 3-4 % de nuevas mutaciones, y presencia en 69,9 % de individuos en la edad pediátrica.8
El objetivo del presente artículo fue revisar los aspectos más sobresalientes relacionados con las enfermedades raras, y su repercusión en la infancia con una visión gastroenterológica.
Métodos
Se realizaron búsquedas no estructuradas de publicaciones en español e inglés en PubMed, Google Scholar, Scimago, SciELO, desde enero 2010 hasta agosto 2021. Se usaron los términos: enfermedades raras, conceptualización, prevalencia, epidemiología, medicamentos huérfanos y ética. Se consideraron los aspectos médicos asistenciales, terapéuticos y económicos con una visión gastroenterológica del problema, y se destacó la importancia de la prevalencia, fundamentada en las manifestaciones clínicas de sospecha y de laboratorio, que permitieron realizar estudios endoscópicos, de biopsia, imagenológicos y genéticos, para la detección oportuna de las ER.
Análisis y síntesis de la información
La epidemiología de las ER estudia e investiga su dinámica y repercusión social, determina la frecuencia y tendencias, y entiende las necesidades reales que caracterizan la asistencia sanitaria. Asimismo, aporta enseñanzas sobre la distribución y experiencias en la conducta terapéutica de los llamados medicamentos huérfanos.9) Además, la epidemiología apoya la ejecución de programas de control y prevención de problemas sanitarios en las poblaciones, y establece las relaciones de gestión entre los sistemas salud-sociedad.10,11
La interpretación de la baja prevalencia varía entre los países. La Unión Europea postula una prevalencia de 5 casos/ 10 000 o 1/ 2000 habitantes. mientras que en EE. UU. se emplea la cifra global de < 200 000 o 40-50 casos por/ 100 000 habitantes del país. En Rusia la prevalencia máxima resulta en 1/ 10 000,12 en Japón < 2/ 10 000 habitantes. En Taiwán el criterio corresponde a 1/ 10 000 habitantes, en Australia, menos 1/ 2000 personas;8 en Turquía no más de 1/ 100 000 habitantes (50 veces menos que la definición de la Unión Europea) y en otros países < 4/ 10 000 habitantes.7) En México se considera rara cuando corresponde a 5/ 10 000 personas. En general, el índice mínimo de baja prevalencia aceptado para ER resultaba en 1/ 10 000 habitantes (tabla1).
En años recientes surgió el término de enfermedades ultra raras con una relación de < 5/ 100 000 habitantes.13
Tabla 1 Prevalencia de enfermedades raras en diferentes países
| País |
Prevalencia por 10 000 habitantes |
|---|---|
| EE. UU. | 6,4 |
| Unión Europea | 5,0 |
| Canadá | 5,0 |
| México | 5,0 |
| Australia | < 5,0 |
| Japón | 4,0 |
| Corea del Sur | 4,0 |
| Rusia | 1,0 |
Fuente: Verma Sub-Committee Report ‘Guidelines for Therapy and
Resulta de interés la introducción por Francia de una definición extra, el término de “cáncer raro”, cuya incidencia corresponde a < 6/ 100 000/ año, y requiere tratamiento especializado por tratarse de un tumor con localización atípica o una enfermedad con características complejas.14 Asimismo, en Escocia se introdujo en 2018 una nueva definición: los medicamentos ultra huérfanos para aquellos que se usan en una condición de prevalencia de 1/ 50 000 o menos, y una proporción muy limitada de enfermos.7,15) De lo anterior, se afirma que una enfermedad se considera rara o huérfana cuando su frecuencia, incidencia o prevalencia resulta baja o en extremo baja, según el límite establecido en consonancia con los criterios de cada país, y que puede modificarse con el tiempo.1,16
La elevada frecuencia de afecciones genéticas y anomalías congénitas se vincularon con la aparición de la mayoría de las ER en el niño, sin embargo, la prevalencia resulta mayor en el adulto que en la infancia, debido a que ciertas enfermedades aparecen en edades tardías, aunque la incidencia de enfermedad grave y la alta mortalidad predominan en el niño.17
Las ER resultan numerosas y heterogéneas; su localización geográfica se considera diferente en consonancia con las variaciones individuales de grave impacto para el paciente, la familia, el personal sanitario (médicos y paramédicos), los sistemas nacionales de salud y la sociedad en sí misma; constituyen una carga económica importante e independiente de las condiciones nacionales y de la propia distribución demográfica.18
En algunos países se establecer dos terminologías: las ER o huérfanas y las enfermedades catastróficas, con diferentes significados. Las ER corresponden a aquellas que relacionan el concepto con su prevalencia o incidencia, y el término “catastrófico” se reserva para aquellas que muestran una fuerte repercusión por el alto costo de los tratamientos necesarios, que sobrecargan a los que las padecen, a sus familias y a los sistemas de salud.8,19).En ambos casos, se considera el criterio predeterminado en distintos países, y en muchos se enumera cuales enfermedades corresponden a cada tipo. Por ejemplo, en Perú las ER incluidas llegan a 399 y las catastróficas ascienden a 16 afecciones, de las cuales 7 pertenecen a enfermedades causantes de los más frecuentes tumores malignos.19
El surgimiento de las denominadas “ciencias ómicas” entre las que sobresale la genómica, resulta decisivo para la comprensión de las ER.20 El desarrollo de las técnicas moleculares permite el estudio de las mutaciones específicas, como en la fibrosis quística, entre otras muchas afecciones.21,22) El desarrollo de las técnicas de secuenciación para el genoma humano permiten el estudio de factores genéticos determinantes con los cuales se logran efectivos diagnósticos, acciones de prevención y participación en el tratamiento; elementos útiles para el establecimiento de las bases para una medicina de precisión en las ER.2
Se plantea que las enfermedades genéticas se deben, entre otros, a mutaciones genómicas, alteraciones cromosómicas o cambios epigenéticos, que se localizan en todas las células de un solo sujeto, o en un mosaicismo, es decir, en porcentaje de células.23
El reconocimiento de las variaciones genéticas en los estudios del genoma humano resultan de gran utilidad para conocer los datos fenotípicos, las características físicas, antecedentes clínicos, y otros elementos de identificación que se vinculan con variantes en el genoma.24
En tiempos recientes, se postula que los próximos 20 años podrían resultar promisorios para los estudios de la secuencia de la genómica humana, para el establecimiento de colaboraciones en la investigación de las ER, y en la ingente necesidad de equidad para aplicar los resultados en la asistencia médica a toda la población necesitada.23
Dilema del diagnóstico
Estudio realizado en 12 000 pacientes de 17 países de la Unión Europea, todos con sistemas nacionales adecuados para la atención en salud, registra que el 25 % de los casos con ER demoran entre 5 y 30 años, desde la aparición de los síntomas iniciales hasta el diagnóstico definitivo. El 43 % obtiene un diagnóstico inicialmente equivocado, de estos, 79 % recibe tratamiento inapropiado y, entre ellos, el 16 % cirugías innecesarias.25
El 69 % de lo que obtuvieron un diagnóstico tardío, padecen consecuencias desfavorables (secuelas físicas y psicológicas, conductas inapropiados por parte de los padres hacia sus hijos o nacimiento de otro hijo con problemas similares).25)
Los casos de enfermedades con herencia recesiva muestran una recurrencia de 25 %, los de herencia dominante alcanzan una repetición de 50 % en cada nuevo embarazo.26) A lo anterior, se unen las prohibiciones establecidas en distintos países sobre la posibilidad de interrumpir un embarazo por razones genéticas. Además, en el momento del diagnóstico se visibiliza la falta de un adecuado tratamiento, en extremo costoso o no existente en ocasiones.2,26
En las ER no diagnosticadas, se postulan diferentes y supuestas particularidades clínicas:27
Diagnóstico tardío: el dictamen se retrasa, aunque se obtiene por medios relativamente simples o comunes en la práctica clínica. En tal caso existe la posibilidad de que el paciente no se deriva hacia el médicos o equipo especializado.
Diagnóstico complejo: fenotipo no orientador, biomarcadores insuficientes y perfiles genómicos no concluyentes; el paciente requiere de equipo especializado y evaluación en un centro con experiencia o en una red nacional de referencia.
Diagnóstico no solucionado a pesar de que todas las investigaciones disponibles se realizaron en el entorno clínico de expertos: en este caso, el paciente, los médicos y expertos pudieran enfrentarse a un nuevo trastorno no descrito.
La orientación diagnostica desde el primer nivel de atención, resulta un factor de trascendencia para las ER. Con frecuencia, los retrasos referidos en el diagnóstico o su inexactitud, se deben, entre otros, a la afectación de múltiples sistemas o a la fragmentación de la atención en varias especialidades médicas, lo que cual redunda en mayor tiempo para arribar a la identificación de la enfermedad poco común.
La importancia del diagnóstico preciso y precoz, y el asesoramiento genético, repercuten en el desarrollo de la enfermedad, su pronóstico y prevención, y evita condiciones excluyentes en el ámbito social.28
Se plantean suficientes criterios sobre el diagnóstico erróneo, insuficiente o tardío de las ER.29) En esta dirección, se postula que la identificación y el diagnóstico preciso de las ER se relaciona con el acceso a los centros médicos de nivel terciario, en los cuales médicos expertos comunican una adecuada conducta sobre la enfermedad. Asimismo, la distancia y acceso a dichas instituciones de las poblaciones rurales y marginales, aisladas en los países en vías de desarrollo, unido a la carencia de asistencia sanitaria especializada, intervienen como factores determinantes entre las limitaciones para una atención integral, y el logro de un diagnóstico y tratamiento precoces.30
Enfermedades raras en la infancia
La base de datos Orphanet (Unión Europea), auspiciada por el Instituto de Investigación para la Salud y la Investigación Médica (INSERM) de Francia, recoge la mayor cantidad de información acumulada acerca de las investigaciones epidemiológicas de las ER, con precisión y disponibilidad a escala global. Lo anterior significa un aporte decisivo para el conocimiento de las ER, en especial en la infancia,7 en la que el 30 % de estas enfermedades se manifestaban antes de los 5 años.8
Las ER se clasifican en cuatro categorías: letales; graves, pero no letales; manejables y con opciones de tratamiento eficaz. La preparación de un listado con esta clasificación resulta una actividad de elevada complejidad. Su realización encierra un interés poblacional, en el que la consanguinidad juega un rol decisivo como factor de influencia. En este sentido, se plantea la práctica de la endogamia en muchas poblaciones y comunidades indígenas en la región suramericana. Los argumentos para la incorporación de las ER en los listados nacionales se realizan con el consentimiento de los territorios y países, tal como se realiza en España y sus regiones, con una progresiva implementación. Un registro de esta naturaleza puede variar en el tiempo y mostrar flexibilidad.31
Distintos países desarrollaron un estudio geográfico de las ER que resulta de interés para el reconocimiento y la incidencia hereditaria.9,30
En muchas ocasiones, el diagnóstico de la ER no se logra en una primera consulta médica, a veces, por manifestaciones clínicas inespecíficas, por requerir estudios especializados para una identificación precisa, consultas con expertos por afectación de órganos de distintos sistemas, o para evitar errores; todo ello en el ámbito de una situación familiar emocional de preocupación y tensión.
Luego del diagnóstico, se plantea el desafío de la conducta terapéutica a seguir, debido a la indicación de medicamentos en extremos caros, la posibilidad de su acceso, la falta de un tratamiento adecuado o su carencia específica.32) Resulta clásico, en el contexto de las diferentes manifestaciones clínicas, el hallazgo de variaciones en dependencia de los sistemas afectados.
En la edad pediátrica, se orienta en un paciente la identificación diagnóstica, principalmente, por la aparición de síntomas y signos físicos vinculados con alteraciones en el sistema nervioso central, visceromegalia (hepatomegalia, esplenomegalia), malformaciones esqueléticas, enfermedades oculares, cardiovasculares, trastornos respiratorios a repetición; alteración en el crecimiento, en la actividad psicomotora y otras deficiencias en el desarrollo, como la expresión del hablar o el caminar, entre las manifestaciones más sobresalientes del cuadro clínico, y que se relacionan habitualmente con el sistema afectado o con la sintomatología de mayor especificidad del órgano correspondiente.32
El diagnóstico definitivo de una ER necesita, en una fase inicial, la determinación de análisis generales de laboratorio, exámenes de enzimas presumiblemente deficientes, estudios imagenológicos, y la consulta con el genetista, quien se encarga de las investigaciones moleculares, si se sospecha una enfermedad genética, como sucede en casi el 80 % de los casos. En las enfermedades del sistema digestivo, se suman a los elementos mencionados, los estudios endoscópicos con la obtención de biopsia para evaluación histomorfológica, imprescindible como complemento del diagnóstico clínico.
Las ER más prevalentes, causantes de mayor mortalidad, conforman aquellas que afectan el sistema nervioso, las malformaciones congénitas, las enfermedades hematológicas y los trastornos metabólicos en general, con limitada supervivencia debido a que los pacientes no sobrepasan los 5 años de edad.33)
Reconocidas enfermedades raras que afectan los sistemas digestivos, hematológicos, inmuno-reumatológicos, respiratorios y neurológicos; presentan el rasgo, en muchas, de denominarse con el nombre de quienes las describieron. Además, se enfatiza en las que se producen por almacenamiento, que pueden dañar algún órgano en diferentes sistemas. Se muestran ejemplos de enfermedades raras en la infancia, agrupadas por diferentes sistemas y afecciones33) (cuadro 1).
Cuadro 1 Enfermedades raras en la infancia
| Clasificación | Trastornos o afecciones |
|---|---|
| Hematológicas |
Anomalías coagulación sanguínea Hemofilia Deficiencia del factor XII Enfermedades de von Willebrand Histiocitosis de células de Langerhans |
| Inmuno-reumatológicas |
Artritis reumatoide juvenil Síndrome de Sjögren Granulomatosis de Wegener |
| Respiratorias | Fibrosis quística |
| Neurológicas |
Síndrome de San Filippo Síndrome de X frágil Síndrome de Moebius Síndrome de Rett Síndrome de Gilles de la Tourette Síndrome de Asperger Atrofia muscular espinal infantil* Esclerosis lateral amiotrófica |
| Almacenamiento |
Glucogenosis Enfermedad de Gaucher Mucopolisacaridosis |
| Músculo-esqueléticas y conectivas |
Osteogénesis imperfecta Síndrome de Marfán Síndrome de Apert |
| Oncológicas |
Enfermedad de Hodgkin Tumor de Wilms Sarcoma de Ewing |
| Otras |
Síndrome de Hutchinson-Gilford Progeria Insensibilidad congénita al dolor y la anhidrosis |
Fuente: Kliegman y otros;33Ross y otros;34) López-Cortés y otros.35
Es de interés referir que para las atrofias musculares espinales infantiles, se produce un medicamento huérfano, el onasemnogen abeparvovec (comercializado como Zolgensma), fundamentado en terapia génica y que puede ayudar a contrarrestar los efectos de la enfermedad. Se evalúa como el más costoso fármaco a nivel mundial, con excesivo precio de 2,1 millones de dólares.34
Visión gastroenterológica
Las enfermedades digestivas en general representan habitualmente el 20 % de las consultas médicas ambulatorias en la infancia y la adolescencia.36,37) En el contexto de la evaluación de las ER resulta fundamental precisar la incidencia por año y prevalencia sobre los criterios establecidos para catalogar una afección como poco común, lo que en ocasiones podía presentar variaciones según países o desencadenar discrepancias. Por ejemplo, la enfermedad celíaca, constituye un trastorno sistémico autoinmune causado por la ingestión de gluten en individuos generalmente susceptibles, y caracterizada por una variable combinación de manifestaciones clínicas, con anticuerpos específicos y fenotipos HLA DQ2 o DQ8, con enteropatía. La prevalencia de datos más recientes notifica el 1 % en los países occidentales, con un rango entre 0,5-1,2 %,38) y resulta tradicionalmente evaluada como rara en la región andina sudamericana (Colombia, Ecuador, Perú y Bolivia) y en Centro América. Sin embargo, en años recientes en Cali, Colombia, Velasco-Benítez demuestra su presencia en estudios de pesquisa en niños con diabetes mellitus tipo 1, síndrome Down y dolor abdominal crónico, incluso con mayor prevalencia de la enfermedad que en otros países de la región latinoamericana.39,40
Por otro lado, Parra-Medina en 2015, postula que la enfermedad celíaca resulta una afección poco común entre los colombianos.40 Esta discrepancia plantea la importancia de realizar cribados en la infancia mediante estudios epidemiológicos para dilucidar las contradicciones sobre el diagnóstico poco común.
Lo anterior reafirma que los criterios de rareza deben avalarse por pesquisas que demuestren la prevalencia de una afección, y sustenten los criterios establecidos acerca de la enfermedad.
En general, la prevalencia de la enfermedad celíaca en los países andinos de Sudamérica no se conocía con exactitud, pero los estudios globales realizados en los últimos años señalan un rango entre 0,46-0,64 % para toda la región suramericana. Los resultados en México resultan interesantes: de 0,7-2,7 %, país en que se considera tradicional la cultura alimentaria con maíz, que al igual que el resto de los cereales, contiene su propio tipo de gluten. Lo anterior respalda la trascendencia de la difusión del conocimiento sobre las posibles ER y el rigor de búsqueda necesario para definir dicha condición.41,42)
Entre las enfermedades digestivas en el lactante, resultaban importantes las diarreas crónicas en las primeras semanas consecutiva al nacimiento y hasta los tres a seis primeros meses de vida. Si la diarrea no presenta causa infecciosa, puede corresponderse con las denominadas enteropatías congénitas, enfermedades poco reconocidas debido a una serie de trastornos congénitos raros, no diagnosticados habitualmente, que pueden semejar enfermos en estado crítico por enfermedades comunes. Los rasgos de un síndrome de diarrea intratable inducida por la dieta, de tipo osmótica, o relacionada con el transporte de electrolitos, tipo secretora, con antecedente familiar de consanguinidad, orientan el diagnóstico por defecto en las células epiteliales del intestino producidas por distintas categorías de enfermedades, en dependencia del daño intestinal. La identificación de la causa por biopsia de duodeno obtenida por endoscopia, realizada con prontitud, permite en la mayoría de los casos arribar a un diagnóstico preciso.43
La identificación temprana, para una clasificación específica de estas causas congénitas, como la enteropatía en penacho, la inclusión de vellosidades intestinales, defectos de enzimas epiteliales en el transporte de electrolitos (diarrea congénita clorada, diarrea de sodio congénita) y desregulación por autoinmunidad, resultaría decisivo para la salud y la vida del lactante, por tratarse de afecciones graves con evolución tortuosa.43
El desarrollo de los estudios endoscópicos contribuyeron a variar criterios sobre afecciones evaluadas como raras en la infancia, ente ellas, las enfermedades inflamatorias intestinales (EII) -colitis ulcerosa y enfermedad de Crohn-, poco comunes en décadas precedentes, comenzaron a presentarse de forma frecuente, con un aumento progresivo en el diagnóstico, 30 %, en la edad pediátrica, variaciones geográficas con mayor frecuencia en los países industrializados, con formas clínicas particulares que la diferenciaban de la presentación en el adulto. Los estudios de colonoscopia y biopsia intestinal demuestran las lesiones más características de colitis causales de diarrea crónica, rectorragia y dolor abdominal, aunque en muchas latitudes se catalogan en el niño y adolescente como raras enfermedades gastroenterológicas.44,45
Asimismo, se distingue la enfermedad eosinofílica digestiva (esofagitis, gastritis, gastroenteritis, que varía en la infancia, en el curso del Nuevo Milenio, con un incremento anual en su incidencia y prevalencia debido a un mayor conocimiento de la enfermedad.46) El diagnóstico de la enfermedad se diferencia por países,47) y se favorece con una mayor disposición e indicación de los procedimientos endoscópicos en pediatría48) y con el aumento de las afecciones atópicas, aunque en general, en el niño menor y adolescente en muchos países, se mantiene registrada como una ER.48,49
La colitis microscópica, alteración inflamatoria colónica, se consideró, hasta la década de los años 90 del siglo xx, una ER de la infancia, pero en los últimos años un mayor número de estudios evaluaron su incidencia y prevalencia, y se observaron importantes variaciones geográficas. El diagnóstico se basa en una sintomatología compatible con diarrea crónica acuosa y predominio de aspecto macroscópico de normalidad de la mucosa en el examen colonoscópico (80 %) o, en ocasiones, alteraciones inespecíficas de eritema, edema de la mucosa del colon, y criterios histomorfológicos definidos de colitis linfocítica (CL), debido a un incremento de linfocitos intraepiteliales (> 20 linfocitos/ 100 células epiteliales).
En otras ocasiones, el diagnóstico se orienta hacia una colitis colagenosa (CC), caracterizada por una banda de colágeno subepitelial, que por definición mide más de 10 micras (10 μm) de grosor. Estos tipos no se establecieron por las manifestaciones clínicas, resultan similares, y se distinguen por la presencia o ausencia de engrosamiento subepitelial. La CC no ocurre en la edad pediátrica y adolescente, solo se describe en el adulto con edad por encima de 50 años (64/ 100 000).50
La colitis pseudomembranosa (colitis asociada a antibióticos) resulta otra rara afección de la infancia, generalmente relacionada con diarrea por antibiótico y debido a una proliferación excesiva de la bacteria Clostridioides difficile (C difficile), principal causa de diarrea relacionada con los cuidados sanitarios en hospitales estadounidenses. Las manifestaciones clínicas de la infección varían desde colonización asintomática, diarrea leve, hasta síntomas severos de colitis fulminante, con distintos grados de lesión con exudados en forma pseudomembranosa, y en ocasiones megacolon tóxico (toxinas A y B). Su incidencia resulta menor en el niño que en el adulto, pero en los dos últimos decenios se registra un aumento de su prevalencia por su identificación diagnóstica mediante colonoscopia.51,52
Entre las ER digestivas por daño del hígado en el lactante y niño mayor, merecen destacarse el síndrome de colestasis ductular biliar sindromática, descrito por Alagille en 1972, en Francia, (síndrome de Alagille o displasia arteriohepática). Resulta la más importante entre las causas genéticas de colestasis crónica, que cursa con ictericia, prurito severo, xantomatosis, hepatomegalia, facie peculiar; y alteraciones cardiovasculares (soplo sistólico por hipoplasia distal de la arteria pulmonar), óseas (vértebras en forma de alas de mariposa) y oculares (embritoxon posterior), entre los elementos esenciales que conforman el síndrome, debido a alteración en el cromosoma 20.53)
La EW, enfermedad hereditaria (autosómica recesiva) poco conocida, que puede cursar con daño irreparable del hígado; comprende un trastorno del metabolismo del cobre, provocado por alteraciones moleculares del gen atp7b, del cual se informan más de 500 mutaciones genéticas. Las formas de presentación de la EW varían: hepáticas (hepatitis crónica, cirrosis e hipertensión portal); oculares (anillo de Kayser-Fleischer), y neurológicas y psiquiátricas, en dependencia del órgano depósito del cobre. Su diagnóstico precoz resulta importante porque se reconoce como una enfermedad genética tratable, cuya prevalencia estimada más aceptada se ubica en 1/ 30 000, aunque estudios de cribado sugieren un prevalencia mayor, como en el Reino Unido (con una estimada prevalencia clínica menor que la prevalencia genética), y en países asiáticos (Japón 1/ 14 000), con variaciones exponentes de elevadas tasas de consanguinidad en poblaciones con elevado porcentaje de heterocigotos, como en la isla de Creta (1 por cada 15 nacimientos) y la cuenca del mediterráneo.54
El trasplante hepático resultaba una indicación terapéutica para ciertas enfermedades raras con daño severo por cirrosis del órgano, como el síndrome de Alagille, EW, tirosinemia tipo b y afecciones por depósito de glucógeno (tipo I, III y IV), enfermedades que cursan con riesgo de carcinoma hepatocelular o cirrosis con progresiva insuficiencia o fallo hepático.55).
La colangitis esclerosante primaria, se considera otra rara enfermedad hepática crónica descrita en 1960, caracterizada por estenosis multifocales de las vías biliares por inflamación y fibrosis, con progresivo daño del hígado; presenta estrecho vínculo con las EII en la mayoría de los casos.56 El diagnóstico se basa en hallazgos colangiográficos; aumentó paulatinamente en adultos en EE. UU. y países europeos, y en años recientes, se diagnostica en niños, por el advenimiento de la resonancia magnética colangiográfica versus la colangiografía retrógrada endoscópica. La incidencia en el adulto: 0,4 a 2,0 por 100 000/ año; en pediatría es aún más rara, pues se describen muy pocos casos.57).
Las malformaciones congénitas resultaron infrecuentes y se relacionan con factores genéticos como las anomalías cromosómicas y las mutaciones genéticas, sin embargo, la prevalencia e incidencia en conjunto no resulta representativas de ER.
En el tubo digestivo, la atresia esofágica representa la malformación más frecuente, con una prevalencia de 1/ 500 nacidos vivos, en la que hasta 50 % de los casos se asocian con otras malformaciones. Asimismo, la atresia intestinal resulta otra causa grave de obstrucción intestinal en el recién nacido, producida la mayoría de las veces por necrosis isquémica del intestino fetal.58
Resulta de interés resaltar el surgimiento de nuevas enfermedades raras por intolerancia digestiva como la intolerancia a la histamina (IH). Evaluada como enfermedad controvertida, poco descrita en la infancia, con manifestaciones clínicas digestiva y extradigestiva. Presenta respuesta positiva a la dieta baja en histamina y la disminución de las concentraciones de la diaminooxidasa plasmática la respalda.59
La enfermedad de Whipple, resulta una infección sistémica grave, rara en el adulto, de trasmisión fecal-oral causada por la actinobacteria Tropheryma whipplei que afecta el intestino delgado y causa absorción intestinal severa; en ocasiones daña otros órganos, como el sistema nervioso central o el corazón.60 En el niño, en la actualidad, se redescribe con variaciones clínicas y autolimitadas, infradiagnósticada, y que se manifiesta como gastroenteritis aguda infecciosa.61 La enfermedad se documenta en 15 % de los casos de diarrea aguda en niños africanos aunque en el sudeste asiático, como en Laos, la prevalencia resulta alta62) y se asocia con otros síntomas como fiebre, tos y trastorno del sueño; a diferencia de la forma clásica en el adulto con presencia de diarrea crónica, artralgia y pérdida de peso. El tratamiento consiste en antibioticoterapia (cefalosporinas (ceftriaxone( por dos semanas, seguido de trimetropim-sulfametoxazol durante 1 año), aunque existen otros tratamientos alternativos. La enfermedad de Whipple se evalúa como enfermedad emergente.60
Se muestran las más reconocidas ER del sistema digestivo en la infancia distribuidas por órganos (cuadro 2).
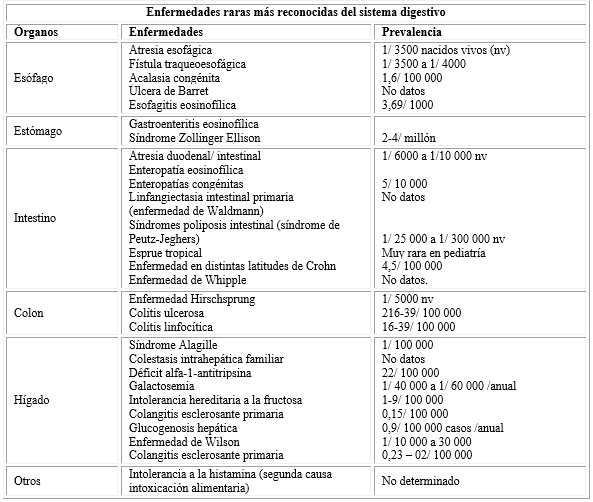
Tratamiento de las enfermedades raras
Se establecieron distintos procedimientos:
Conducta alimentaria apropiada, con el propósito de realizar control estricto en la dieta que elimine el efecto nocivo de determinados alimentos, como sucede en la fenilcetonuria, galactosemia, malabsorción de glucosa-galactosa y otras.
Procederes con hormonas o enzimas, para sustituir las deficiencias, como las enzimas pancreáticas en la fibrosis quística o el bifosfonato en la osteogénesis imperfecta.
Medicamentos huérfanos.
El otro desafío: el tratamiento con medicamentos huérfanos
Los medicamentos denominados huérfanos resultan clave para llamar la atención acerca de la trascendencia de las ER y sobre los tratamientos, por los costosos precios o su carencia, definidos por instituciones de EE. UU. y a posteriori, en países de la Unión Europea. Los nuevos requerimientos terapéuticos para las distintas formas de expresión de las ER permitieron, en los últimos 40 años, disponer de diferentes medicamentos para dichas afecciones, aunque en muchas el efecto resultaba sintomático, por la inexistencia de una terapia específica.63
La aparición de noveles tratamientos para regular los eventos en la fisiopatología de las ER, comprenden una modalidad de terapias costosas por la necesidad de mantener la indicación a largo plazo. Los denominados “potencializadores”, para la fibrosis quística constituyen un ejemplo en esta dirección.64
El auge de estos tratamientos se manifiesta en las ganancias obtenidas por la industria farmacéutica que oscilan entre 6-10 % anual, y en el incremento de los precios de estos nuevos productos que, además, encarecen la atención de las ER.
Los montos exorbitantes de algunos fármacos expresan el drama de los tratamientos impagables, como es el caso mencionado de la medicación para la atrofia muscular espinal infantil.
Es preciso enfatizar que los procederes terapéuticos resultan en extremo costoso (con medicamentos a un precio extraordinariamente elevado, hasta un millón de dólares), como el referido fármaco onasemnogen abeparvovec (Zolgensma, nombre comercial), cuya patente pertenece solo al laboratorio Novartis, con valor que duplicó el de 850 000 dólares del fármaco voretigén neparvovec (Luxturna, nombre comercial), anterior fármaco de mayor costo existente, usado para la ceguera causada por una enfermedad hereditaria rara.5,65
El universo, en el Nuevo Milenio, requiere con emergencia de un orden nuevo en la gestión de los medicamentos huérfanos para la supervivencia de pacientes con ER; con un enfoque global fundamentado en principios humanos y éticos, frente a un mundo en el cual los gastos militares resultan superiores a la inversión dedicada a las investigaciones científicas, como la producción de nuevos fármacos y su aplicación en tratamientos, hoy de un alto costo, como ocurre en las ER.63,65
Es importante enfatizar que los resultados de las investigaciones en ER, en los últimos años, aumentan la supervivencia en muchas de ellas, pero, la producción de nuevas terapias y de tratamientos costosos, no rentables resultaba aún limitada para múltiples afecciones, y la posibilidad de su adquisición se manifiesta como expresión de inequidad social, y de crisis de elementales principios éticos por la vida humana.31
Conclusiones
Las enfermedades raras constituyen afecciones severas con gran discapacidad para el enfermo. En conjunto representan un importante problema sanitario y social, de impacto en la edad pediátrica con repercusión para la familia y la sociedad.
La OMS estima que, en su mayoría, las ER corresponden a causas genéticas y se relacionan con un elevado porcentaje de endogamia, con el rasgo de poco habituales en unos países, mientras se manifiestan con frecuencia en otros, y no se catalogan raras. La prevalencia se desconoce en muchos países, no abundan estudios sobre la existencia global de las ER unido al drama acerca del significado de los medicamentos huérfanos, específicos para el tratamiento de diferentes afecciones, que representa un reto social para la salud pública.
En el contexto de la visión gastroenterológica sobresalen diferentes enfermedades del hígado y del intestino, con elevado progreso diagnóstico basado en los estudios endoscópicos y de biopsia, en especial en el lactante y el niño de corta edad, contribuciones decisivas que variaron criterios sobre afecciones del tracto gastrointestinal reconocidas antaño raras, pero que comenzaron a ser más diagnosticadas.

