










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Obstetricia y Ginecología
versión On-line ISSN 1561-3062
Rev Cubana Obstet Ginecol v.31 n.1 Ciudad de la Habana ene.-abr. 2005
Instituto nacional de oncología y radiobiología.
Infección por el virus del papiloma humano y factores relacionados con la actividad sexual en la génesis del cáncer de cuello uterino
Lic.Grettell León Cruz1 y Dr. Omar de Jesús Bosques Diego2
Resumen
El virus del papiloma humano (VPH) es el principal agente etiológico infeccioso asociado con la patogénesis del cáncer de cuello uterino. Se plantea que el conocimiento de la virología y las manifestaciones clínicas de este virus constituyen el eslabón fundamental en el entendimiento del proceso neoplásico. Los estudios epidemiológicos de las lesiones premalignas del cuello uterino han demostrado una fuerte asociación entre la práctica sexual y la aparición de tumores malignos. Se indica que las mujeres con múltiples patrones sexuales, embarazos e interrupciones a temprana a edad e historias de infecciones, aumentan el riesgo de padecer la enfermedad.
Palabras clave: Cáncer cervico-uterino, infección por virus papiloma humano, conducta sexual.
El cáncer de cuello uterino es el resultado de la progresión de leves anomalías epiteliales llamadas displasias o neoplasias intraepiteliales (NIC), diagnóstico frecuente en mujeres entre los 20 y 30 años de edad, pasando por carcinoma in situ, entre los 25 y 35 años, a carcinoma invasivo en mujeres mayores de 40 años.1 Los tumores malignos del cuello uterino en estadios tempranos son claramente identificables por la confirmación histoanatomopatológica, sin embargo, las cifras de incidencia de esta enfermedad continúan alarmando. Aproximadamente la mitad del total de las mujeres que desarrollan cáncer de cuello uterino invasivo mueren después de los 5 años de diagnosticadas.2
En los últimos años se han logrado importantes progresos en el estudio de la (s) causa (s) de la aparición del cáncer de cuello uterino. Actualmente es aceptado como principal causa la infección por VPH de alto riesgo y sus precursores,3 sin embargo, estudios epidemiológicos han mostrado que sólo una pequeña fracción de mujeres infestadas con VPH eventualmente progresan a lesiones intraepiteliales de alto riesgo y carcinoma in situ,4 por lo que se ha asumido que otros factores actúan en conjunto con el VPH, influenciando el riesgo de transición de la infección VPH cervical a malignidades.4
La promiscuidad, sin ser sinónimo de cáncer de cérvix, constituye un importante factor de riesgo. Numerosos mecanismos han sido sugeridos para explicar la relación entre el riesgo de padecer la enfermedad y los diversos elementos asociados con las relaciones sexuales, entre ellos la edad del comienzo de las relaciones sexuales y la transmisión de agentes infecciosos (Trichomonas, Gardnerella, Herpes y Virus tipo II (HSV-2).5-7
Virus del papiloma humano (VPH)
Los virus oncogénicos desempeñan un papel etiológico de extrema importancia en varios de los tumores malignos que afectan al hombre.8 Como fue anteriormente mencionado, el VPH ha sido identificado como el factor etiológico fundamental en el desarrollo del cáncer de cuello uterino. En el 90-100 % de los casos diagnosticados con cáncer cervicouterino se ha identificado el ADN transcrito y los productos proteicos de este virus, con una prevalencia del 5-20 %.9 Han sido aislados, secuenciados y clonados al menos 100 tipos, y de ellos, 50 están asociados con el tracto genital femenino.10 Este virus ha sido clasificado según el grado de transformación maligna que ocasiona en la célula infectada. en VPH de alto riesgo y/o de bajo riesgo
Los VPH comprenden un grupo de virus no envueltos, de ADN pequeño, con simetría icosaédrica, que inducen verrugas o papillomas en una gran variedad de vertebrados superiores, incluyendo al hombre. Cada tipo es asociado preferentemente con una lesión clínica especifica y con un sitio anatómico preferencial por cada epitelio escamoso, mucosal o cutáneo.
Entre los más comunes que representan al grupo de bajo riesgo se incluyen los tipos 6 y 11 que usualmente causan verrugas benignas y que ocasionalmente, se asocian con lesiones no invasivas; mientras que los tipos VPH-16 y VPH-18, se corresponden con los de "alto riesgo" por su gran potencial carcinogénico. El VPH-16 es el tipo que aparece, fundamentalmente en los tumores invasivos y en los de alto grado de malignidad; el VPH-18 se relaciona con el carcinoma pobremente diferenciado y con un mayor compromiso de los ganglios linfáticos. Tanto el genoma del VPH-18 como el del VPH-16 pueden encontrarse como viriones, integrados en el ADN celular o de forma episomal.10-12
La infección por VPH es inicialmente asintomática y la transmisión puede ocurrir antes de que la expresión del virus se manifieste.13 El epitelio diferenciado es necesario para el completo desarrollo y crecimiento del virus, fenómeno conocido como tropismo celular que es evidenciado por la restricción de funciones de replicación viral. La severidad de traumas o erosiones epiteliales9 y la inducción de hiperplasias epidérmicas antes de la infección, son factores locales importantes que favorecen el crecimiento viral. El proceso de infección ocurre fundamentalmente, a través de receptores de integrinas presentes en las células basales. Sin embargo, la lesión puede ser iniciada por lesiones epiteliales pequeñas, siendo poco el acceso a las células básales, donde produce un amplio espectro de cambios morfológicos una vez infestadas.
En el núcleo de la célula hospedera el virus se replica en una relación 25-50 genoma /células, mediado por la actividad de 4 proteínas multifuncionales: E1/E2, E6 y E7.
- E1/E2: región que generalmente se rompe cuando el genoma viral se integra en el genoma hospedero. La disrupción de E2 libera los promotores virales de las oncoproteínas E6 y E7 e incrementa la expresión de estos genes transformantes.
- E6: oncoproteína que se une al producto génico del gen supresor de tumor: p53 (proteína activada por la fosforilación de proteínas sensibles al daño del ADN), formando un complejo E6-p53 que es blanco posterior para la degradación, y provoca fallos en los mecanismos de proliferación y apoptosis.14
La E6 sintetizada por los VPH-6 y VPH-11 muestran una significativa disminución en la capacidad de unión a la p53, lo que podría explicar la asociación menos transformante de estos para la célula.12-16
- E7: esta oncoproteína promueve la trascripción viral por 2 vías.
- Se une al producto génico del gen del retinoblastoma (Rb), liberándose el factor de transcripción E2F, fundamental en la promoción de la síntesis del ADN, tanto del virus como de la célula (figura 1).
- Se une y activan determinados complejos de ciclinas, como la p33-dependiente de quinasa 2, la cual controla la progresión del ciclo celular. La respuesta de las células infestadas ante este fenómeno es la producción de un inhibidor de ciclina quinasa: la proteína p21cip1, que es transcrita a partir de un ARNm secuestrado que existe en las células basales y parabasales. La proteína p21cip1 es típicamente producida por transcritos estimulados por p53, por lo que, si p53 es inactivada, p21cip1 no puede transcribirse. Interesantemente, gran cantidad de E7 puede unirse y bloquear la actividad del inhibidor. Las cantidades relativas de E7 y p21cip1 determinan cuándo la célula entra en fase S del ciclo celular y replica el ADN viral o cuándo bloquea la producción del virus. La célula donde E7 se une a p21cip1 se convierte en koilocito y produce partículas virales9 (figura 1).
Normalmente la proteína p53 activada es requerida para detener el ciclo celular en la fase G1 como resultado de la estimulación directa a p21cip1. Una ausencia o inhibición de esta propiedad conduce a inestabilidad genómica.12 Alternativamente, cuando los daños en el ADN son más severos o cuando hay una gran replicación viral, p53 puede activar la vía apoptótica (figura 1).
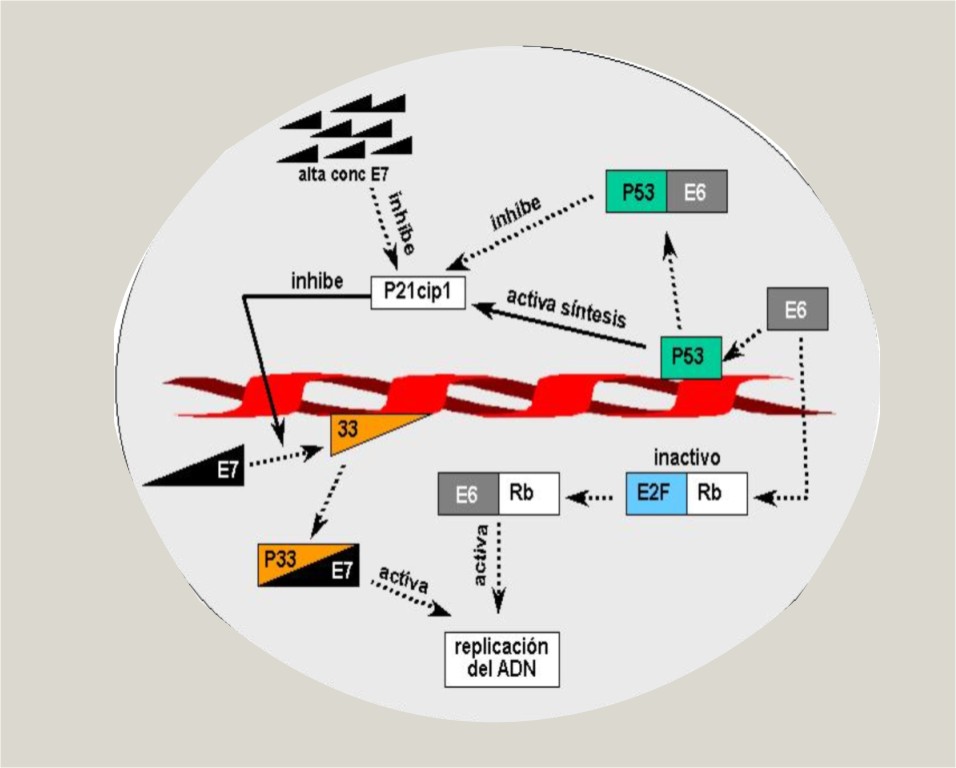
Fig. 1
En las lesiones persistentes, el genoma viral continúa estimulando a las células basales a ignorar el daño en el ADN, que por consiguente, se van acumulando. La estimulación por E6 y E7 de los VPH de alto riesgo produce clones con una larga vida media, pasando al punto conocido como de mortalidad 1 o M1, donde aún las células no son inmortales. Un importante paso en la inmortalización lo constituye la liberación de los telómeros.
Normalmente los telómeros se acortan tras cada generación celular. Cuando alcanzan un tamaño determinado se produce una señal de muerte para las células. El largo y la estabilidad de los telómeros es mantenido por las telomerasas. La oncoproteína E6 puede activar las telomerasas y mutaciones adicionales pueden estabilizar los telómeros, promoviendo el paso de la célula a la fase de mortalidad 2 o M2. No se conocen en detalles cómo mutaciones adicionales e independientes promueven la transición de las células inmortalizadas a células malignas.9
La integración del genoma viral implica, tanto la destrucción del genoma de la célula hospedera como la del propio virus. Las alteraciones genéticas resultantes de la interacción célula-virus están relacionados, directa o indirectamente con la inmortalización de la célula 16 (figura 2).
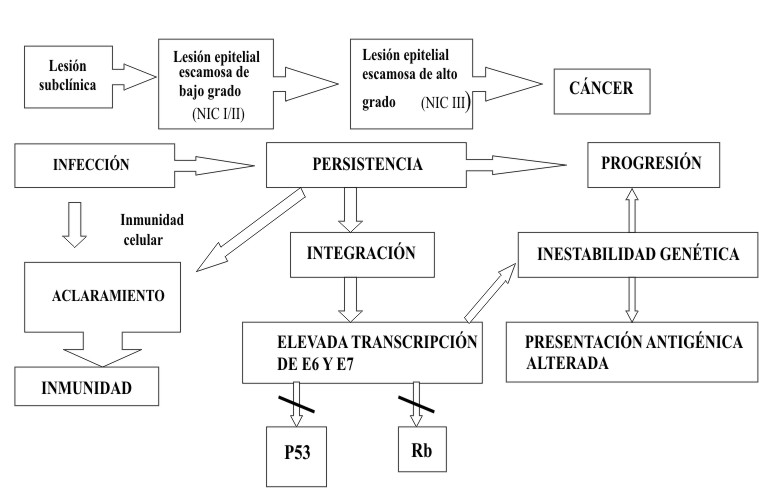
Fig. 2
Después de la infección sólo un pequeño número de genes virales persisten en la membrana basal. Se acumula el ADN del virus en la superficie epitelial con aumento de su replicación y son expresados diferentes mensajeros. Las proteínas virales son sintetizadas y el ADN replicado es empacado en las proteínas de la capsida viral, liberándose así las partículas de infección a la superficie de la epidermis. Todo esto constituye el período de incubación viral, que tiene una duración de 3 a 4 meses.10 Muchas mujeres infestadas no desarrollan signos o síntomas clínicos pero son reservorios del virus por períodos variables de tiempo (Protocol, IARC,1997).
Hipótesis alternativa
Estudios genéticos-moleculares han sugerido 3 mecanismos diferentes para la inducción del cáncer de cérvix:
- Mecanismo relacionado con el VPH: los efectos de E6 y E7 en las proteínas de regulación del ciclo celular de la célula hospedera.
- Consecuencias de la integración viral: impacto específico en los sitios de integración.
- Acumulación de daños genéticos necesarios para el desarrollo del fenotipo que puede o no estar relacionado con el virus.
La existencia de este mecanismo se sustenta en hallazgos de pérdida de heterogocidad.
Los 2 primeros mecanismos han sido explicados el tercero se basa en la existencia de un pequeño porcentaje de casos donde el diagnóstico de carcinoma de cérvix no está relacionado con la infección por VPH. Se han comparado los factores etiológicos descritos para el desarrollo de la afección, tanto en los casos VPH negativos como en los positivos, y existe similitud en edad pico de incidencia, número de patrones sexuales, edad de comienzo de las relaciones sexuales, tiempo de consumo de anticonceptivos orales, paridad, etc., por lo que la explicación a esta hipótesis sólo se sustenta en el hecho de que: 1) las células epiteliales son capaces de desarrollar cáncer y que el cáncer crece en todos los tejidos humano por causa viral o no, así las células del útero también tienen esta capacidad; 2) y/o los genes celulares que están envueltos en la carcinogénesis relacionada con VPH podían ser capaces de generar o inducir espontáneamente mutaciones que dieran lugar al cáncer en ausencia de VPH.
Estos casos son considerados como una verdadera entidad biológica.9
Conducta sexual
La infección por VPH de las células del epitelio cervicouterino es considerada, en términos biológicos, como una enfermedad de transmisión sexual a través del contacto con el epitelio anogenital infestado, poco después de iniciada la relación sexual (Protocol IARC, 1997). El número de parejas sexuales, no es más que el reflejo de la probabilidad de exposición al VPH y demás agentes infecciosos,9 así la vida sexual incrementa la frecuencia del padecimiento de forma importante, sobre todo en aquellas mujeres que la inician antes de los 16 años de edad.
En la adolescencia y durante los primeros embarazos se produce la migración fisiológica de la unión escamocolumnar hacia el endocérvix. En este proceso el epitelio cilíndrico es reemplazado por el epitelio plano estratificado originando la llamada zona de transición, donde la susceptibilidad al riesgo de transformación maligna/célula blanco es probablemente mayor que en cualquier otro tejido sujeto al cáncer.17 Estos cambios son más activos precisamente en etapas tempranas de la vida, donde también la vida sexual es más activa, pero declinan después de la menopausia.
Agentes de transmisión sexual
Las enfermedades ginecológicas pueden afectar el crecimiento de la flora bacterial vaginal.6 Existe una fuerte asociación entre Trichomonas vaginalis y el riesgo de padecer cáncer de cuello uterino (se incrementa en 3 veces).7
Por otra parte, la Gardnerella vaginalis es detectada en el 50 % de las pacientes con tumores malignos del cérvix,lo que sugiere que puede estar fuertemente asociada con el cáncer de cuello uterino.6 El complejo N-cadherina/catenina es un componente estructural importante en la adhesión de las células epiteliales. Bajo determinadas condiciones, la -catenina puede ser liberada de este complejo, luego de lo cual se une a determinados factores de transcripción en el núcleo de las células. Este mecanismo estimula la expresión de genes que regulan la apoptosis y el ciclo celular. En estudios in vitro se ha demostrado que la clamidia rompe el complejo provocando el secuestro de la N-cadherina con la inclusión de la clamidia. Este puede ser el mecanismo por el cual esta última altera la función de las células epiteliales18 y contribuye a la transformación maligna de la célula.
Paridad y edad del primer embarazo
Mujeres portadoras del ADN del VPH, con 7 o más embarazos a término, tienen un riesgo de padecer la enfermedad de 4 veces más que mujeres nuliparas o con menor número de hijos.9 Como se mencionó anteriormente la puerta de entrada del VPH es el epitelio erosionado, lo cual es muy frecuente tras los partos. Sin embargo, sólo en aquellas mujeres con menos de 16 años, donde el epitelio está en fase de transición este es más susceptible a las lesiones. En los embarazos a término y partos naturales la probabilidad de traumas en la zona de transición en el cuello uterino no es frecuente, por lo que la influencia de este factor es cuestionable.
Los estudios relacionados con la etiología de cáncer de cuello uterino han experimentado importantes progresos. Se ha demostrado que la presencia del ADN del VPH y sus precursores es la causa fundamental para el desarrollo, mantenimiento y progresión de las neoplasias malignas del cuello uterino y del cáncer de cérvix, además, que la transmisión sexual es la principal vía de adquisición de este virus.
El cáncer de cuello uterino continúa siendo la segunda causa de muerte por tumores malignos en la mujer, por lo que el seguimiento de la población femenina mediante el programa de prevención precoz con la realización de las citologías vaginales y el testaje del VPH para la clasificación en grupos de riesgos de las mujeres infestadas, debe ser considerado y evaluado como una alternativa de detección primaria.
Summary
Human papilloma virus infection and factors related to sexual activity in the genesis of the cervix uteri cancer
The human papilloma virus (HPV) is the main etiological infectious agent associated with the pathogenesis of cervix uteri cancer. It is stated that the knowledge of virology and the clinical manifestations of this virus are the fundamental link in the understanding of the neoplastic process. The epidemiological studies of the premalignant lesions of the cervix uteri have showed a strong association between sexual practice and the appearance of malignant tumors. It is indicated that women with multiple sexual patterns, pregnancies and abortions at an early age and history of infections, increase the risk of suffering from this disease.
Key words: Cervix uteri cancer, human papiloma virus infection, sexual conduct.
Referencias bibliográficas
1. Louise A, Brinton, Robert NH. Epidemiology of gynecologic cancer. En: Hoskins WJ, Pérez CA. Young RC. Principles and Practice of Ginecologic Oncology. 3ed. Lippincott; Williams and Wilkins; 2000. p. 3-27.
2. González MJ, González B, Biete SA. Ginecología Oncológica. 3ed, Madrid: Menéndez Editores, 2000. p. 141.
3. Bosch FX, Muñoz N, Sanjosé S. Human papillomavirus and other risk factors for cervical cancer. Biomed Pharmather 1997;51:268-75.
4. Castellsague X, Bosch FX, Munoz N. Enviromental co-factors in VPH carcinogenesis. Virus Res 2002;89(2):191-9.
5. Mori M, Sagae S. Recent progress in epidemiologic research of uterine cancer. To Kagaku Ryoho. 2001;28(2):174-8.
6. Mikamo H, Sato Y, Hayasaki Y, Kawazoe K, Izumi Ito K, YamadaY, et al. Intravaginal bacterial flora in patients with uterin cervical cancer. High incidence of detection of Gardnerella vaginalis. J Infect Chemother. 1999;5(2):82-5.
7. Sayedel-Ahl SA, el Wakil HS, Kamel NM, Mahmot MS. A preliminary study on the relationship beteween Trichomonas vaginalis and cervical cancer in Egyptian women. J Egypt Soc Parasitol. 2002;32(1):167-78.
8. Vonka V, Hamsikova E, Sobotkova E, Smahel M, Kitasato H, Sainerova H, et.al. Papillomaviruses and human tumors. Cas Lek Cesk 2000;139(Suppl 1):27-9.
9. Bosch FX, Lorincz A, Muñoz NC, Meijer JLM, Shah KV. The causal relation between human papillomavirus and cervical cancer. J Clin Pathol. 2002;55:244-65.
10. Jastreboff AM, Cymet T. Role of the human papillomavirus in the development of cervical intraepitelial neoplasia and malignancy. Postgrad Med J. 2002;78:225-8.
11. Pillai RM, Lakshmi S, Sreekala S, Devi Ganga T, Jayaprakash PG, Rajalakshmi TN, et al. High-risk Human Papillomavirus infection and E6 protein expression in lesions of the Uterine Cervix. Pathobiology 1998;66:240-6.
12. Van Driel WJ, Kenter GG, Fleuren GJ, Melief CJM, Trimbos BJ. Immunotherapeutic strategies for cervical squamous carcinoma. Curr Therap Issues Gynecologic Cancer. 1999;13(1):259-71.
13. Jastreboff AM, Cymet T. Role of the human papillomavirus in the development of cervical intraepitelial neoplasia and malignancy. Postgrad Med J 2002;78:225-8.
14. Dellas A, Torhorst J, Jian F, Proffitt J. Prognostic value of genetic alterations in invasive cervical squamous cell carcinoma of clinical stage IB detected by comparative genomic hibridization. Cancer Res. 1999;59:3475.
15. Bosch FX, Rohan T, Schneider A, Frazer I, Pfister H, Castellsagué X et al. Papillomavirus research update: highlights of the Barcelona HPV international papillomavirus conference. J Clin Pathol. 2001;54:163-75.
16. Ferenczy A, Jenson B. Tissue effects and host response. The key to the relational triage of cervical neoplasia. Obstet Gynecol Clin North Am 1996;23(4):759-82.
17. Ponten J, Guo Z. Precancer of the human cervix. Cancer Surv 1998;32:201-29.
18. Prozialeck WC, Fay MJ, Lamar PC, Pearson CA, Sigar I, Ramsey KH. Chlamydia trachomatis disrupts N-cadherin-dependent cell-cell junctions and sequesters beta-catenin in human cervical epithelial cells. Infect Immun. 2002;70(5):2605-13.
Recibido: 28 de diciembre de 2004. Aprobado: 10 de mayo de 2005.
Lic. Gretell León Cruz. Instituto Nacional de Oncología y Radiobiología. Ciudad de la Habana, Cuba.
1 Licenciada en Bioquímica
2 Especialista de I grado en Medicina General Integral, Especialista de I grado en Oncología.

