










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Obstetricia y Ginecología
versión On-line ISSN 1561-3062
Rev Cubana Obstet Ginecol vol.42 no.2 Ciudad de la Habana abr.-jun. 2016
REVISIÓN BIBLIOGRÁFICA
Enfermedad de células falciformes en el embarazo
Sickle Cell Disease in Pregnancy
Xiomara Pujadas Rios, Lala Laura Viñals Rodríguez
Hospital General Docente "Dr. Enrique Cabrera". La Habana, Cuba.
RESUMEN
La anemia de células falciformes o drepanocitosis, es una de las hemoglobinopatías estructurales más comunes en el mundo. La clínica se resume en oclusión vascular e isquemia tisular, anemia hemolítica y la susceptibilidad a infecciones. La procreación en mujeres con hemoglobinopatías deviene un grave problema de salud, que exige una atención diferenciada y multidisciplinaria. Para esta afección no existe tratamiento especifico definitivo, el arsenal medico existente solo puede manejar los efectos y no la causa. La siguiente revisión tiene como objetivo ofrecer a los profesionales algunos aspectos relacionados con la fisiopatología, una discusión del problema clínico, diagnóstico y opciones terapéuticas de la enfermedad, lo que permite contribuir en la reducción de la morbilidad y mortalidad materna y perinatal. Se concluye que un alto índice de perspicacia y buen diagnóstico es menester para obtener resultados óptimos en las embarazadas afectadas por enfermedad de células falciformes.
Palabras clave: anemia de células falciformes; drepanocitosis; embarazada; clínica; diagnóstico; prevención.
ABSTRACT
Sickle cell anemia or sickle cell disease is one of the most common structural hemoglobinopathies in the world. The clinic is summarized in vascular occlusion and tissue ischemia, hemolytic anemia and vulnerability to infections. Procreation in women with hemoglobinopathies becomes a serious health problem that requires a differentiated and multidisciplinary care. There is no definitive specific treatment for this condition, the existing medical resources can only address the effects and not the cause. The following review aims to offer professionals some aspects related to the pathophysiology, a discussion of the clinical problem, diagnosis and treatment options, which can contribute in reducing morbidity and maternal and perinatal mortality. It is concluded that high level of insight and good diagnosis are necessary for optimum results in pregnant women affected by sickle cell disease.
Keywords: sickle cell anemia; sickle cell disease; pregnant; clinic; diagnosis; prevention.
INTRODUCCIÓN
La anemia drepanocítica -sickle cell como se denominó originalmente- constituye un descubrimiento relativamente reciente de una alteración de la sangre. No hubo referencias de esta anemia hasta el siglo XX, los primeros hallazgos clínicos fueron reportados por James Bryan Herrick en 1910. En Cuba se conoce comúnmente como sicklemia y al paciente se le denomina sicklémico lo que evidentemente fue una adaptación a nuestro lenguaje para facilitar su identificación popular.
En diciembre de 1904, un paciente llamado Walter Clement Noel ingresó en el Hospital Presbiteriano de Chicago porque aparentemente sufría de anemia. Ernest Irons, interno colaborador con Herrick, investigaba la sangre de Noel cuando notó la irregularidad en la forma de sus glóbulos rojos.
Fue esta la primera vez que se asoció la irregularidad de la forma de los glóbulos rojos y se descubrió la anemia drepanocítica o anemia por células falciformes.
En 1922, Verne Mason denominó a esta enfermedad "sickle-cell anaemia", basado en la descripción de Ernest Irons en lo relacionado con la forma que adoptan los glóbulos rojos. La enfermedad fue también denominada durante algún tiempo Herrick´s syndrome (síndrome de Herrick).1
En 1949 el químico norteamericano Linus Carl Pauling y algunos de sus colegas investigaron sobre la anemia drepanocítica (sickle-cell anaemia) y fueron los primeros en demostrar que la causa de la enfermedad era una anormalidad de la molécula de hemoglobina.2
La alteración genética que determina la Hb SS es consecuencia de una mutación de genes que ocurrió hace miles de años, predominantemente en el continente africano. Allí hubo tres mutaciones independientes en los pueblos del grupo lingüístico Bantú y en los grupos étnicos Benin y Senegal.
Varios investigadores asocian la mutación genética a la respuesta del organismo a la agresión sobre los glóbulos rojos del Plasmodiun falciparum agente etiológico de la malaria. Hace miles de años la prevalencia de la malaria en estas regiones es alta, los portadores del rasgo falciforme adquieren resistencia a esa enfermedad.3
Con la emigración forzada de los pueblos de África a causa de la esclavitud en América, el gen fue difundido a todos los continentes, por lo que en la actualidad constituye la enfermedad genética de carácter mundial más prevaleciente.
En EE. UU. el mestizaje entre los blancos europeos de distintas regiones, con los grupos originarios indígenas y posteriormente con africanos, fue menos intenso; por lo que el gen de la Hb S permanece solo en la comunidad negra.
En Colombia, Brasil, Venezuela y Nicaragua hubo un mestizaje primario de españoles y portugueses con indígenas y posteriormente con negros. En países como México, Ecuador, Guatemala, El Salvador, Panamá, Perú y Bolivia existe una población numéricamente importante de indígenas y mestizos.
Los países con un gran contingente de población negra son: Brasil, Colombia, Cuba, República Dominicana, Haití, Jamaica, Trinidad y Tobago, Antillas Holandesas, Honduras, Surinam y EE. UU., países donde prevalece el gen Hb S en un rango del 6 % al 10 %.4
En África Ecuatorial y Occidental hay prevalencia del gen Hb S en un índice del 40 % y en el área del Mediterráneo, Sur de Italia (Sicilia), Grecia e India, hasta de un 20 %.1,2
La OMS en la 59ª Asamblea Mundial de la Salud,1 decretó la prevalencia de anemia por células falciformes, y encuentra que aproximadamente 5 % de la población es portadora de genes causantes de hemoglobinopatías. Cada año nacen aproximadamente 300 000 niños con hemoglobinopatías importantes, de los cuales más de 200 000 son africanos con anemia falciforme.
Las hemoglobinopatías son consideradas por la Organización Mundial de la Salud (OMS) como "un programa prioritario de salud en el mundo". Según su comportamiento epidemiológico, representan un problema de salud pública a nivel nacional y mundial. En el nivel individual, constituye una necesidad de atención a la salud, dadas las repercusiones e impacto en la vida de las gestantes y el feto.
ASPECTOS GENERALES
FISIOPATOLOGÍA
La anemia de células falciformes es una hemoglobinopatía, enfermedad que afecta la hemoglobina, una proteína que forma parte de los glóbulos rojos y que se encarga del transporte de oxígeno. Es de origen genético y se da por la sustitución de un aminoácido en su conformación.
Es un defecto de herencia autosómica recesiva caracterizado por la presencia de hemoglobina S (Hb S) en el eritrocito. Esta se diferencia de la Hb A en que se intercambia el aminoácido Acido glutámico por Valina en la posición seis de la cadena beta. Esto se atribuye a una mutación en el gen correspondiente de la cadena beta que se haya localizado en el cromosoma once. En el triplete del ADN afectado se encuentra una Adenina en vez de Timina, esto resulta en que el ADN de la Beta globina codifique en el sexto residuo al aminoácido Valina en el lugar del Ácido glutámico.5
El resultado final es el siguiente: una molécula de hemoglobina A, que posee 574 aminoácidos iguales a los de la hemoglobina S en 572 y diferentes de la hemoglobina A en dos aminoácidos. Esta diferencia mínima causa graves trastornos en los pacientes afectados, constituye el principio fisiopatológico de la enfermedad.
En la drepanocitosis dos hechos son importantes, uno es la anemia hemolítica crónica y otro la oclusión vascular. La anemia hemolítica se debe a profundas alteraciones de la membrana que llevan a la destrucción acelerada del hematíe y la oclusión vascular es un proceso complejo en el que participan muchos factores: la deshidratación del glóbulo rojo, alteraciones de su membrana, aumento de su adhesión al endotelio, alteraciones intrínsecas de las células endoteliales y otros.6
La anemia drepanocítica ocasiona varios fenómenos vaso-oclusivos capaces de afectar cualquier órgano de la economía, se caracteriza por la presencia de hematíes en forma de hoz (falciforme) o de media luna en la sangre periférica, que son los responsables de las manifestaciones clínicas y hematológicas de la enfermedad (Fig. 1).
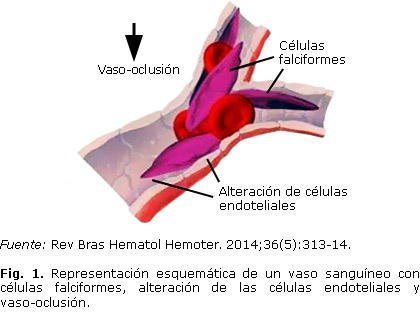
En Cuba, la enfermedad se ha estudiado extensamente, lo cual ha contribuido al conocimiento de sus manifestaciones clínicas y hematológicas; es el único que cuenta con un plan nacional de atención integral que jerarquiza el Instituto de Hematología e Inmunología. Gracias a estos estudios se ha mejorado la calidad de vida del paciente y disminuye, de manera significativa, la morbiletalidad.
La forma más frecuente de drepanocitosis en Cuba, es la anemia drepanocítica (AD) o hemoglobinopatía (SS), le siguen en frecuencia, la hemoglobinopatía SC (Hb SC) y la Sβ talasemia (Sβ tal); esta última puede ser Sβ0 tal o Sβ+ tal. La frecuencia del estado de portador AS es de un 7 % en el mundo y cada año nacen más de 330 000 niños afectados.7
RASGO FALCIFORME (AS)
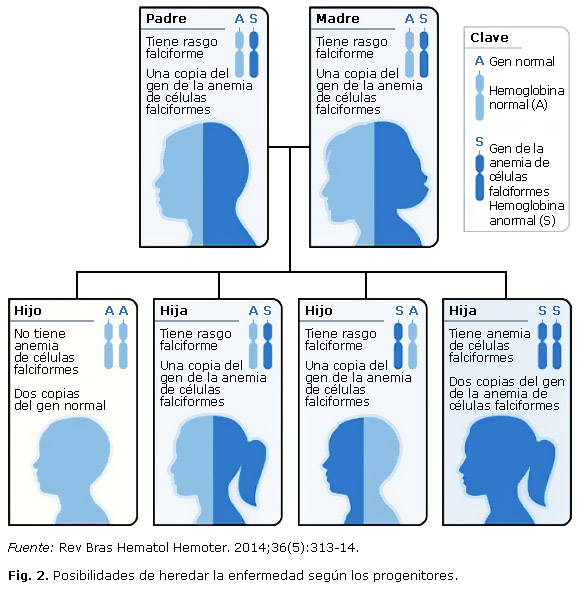
Son personas portadoras de la Hb S asintomáticas, con cifras de Hb, morfología sanguínea y desarrollo físico normal. La concentración de Hb S es menor del 50 %; sin embargo, en algunas circunstancias de anoxia pueden presentar complicaciones. Los heterocigotos Hb (AS) tienen anemia leve y en circunstancias normales presentan la misma eficacia biológica que los homocigotos Hb (AA); con la ventaja de que la Hb falciforme lo protege contra el protozoo de la malaria, fenómeno denominado polimorfismo compensado. La alteración clínica más frecuente es la hipostenuria o hematuria. Se han descrito algunos casos de radomiolisis tras el ejercicio intenso, el rasgo drepanocítico no requiere tratamiento. Cuando ambos padres tienen el rasgo falciforme Hb (AS) en cada embarazo habrá las siguientes posibilidades: 25 % de que el hijo sea normal Hb (AA), 50 % de que el hijo herede la Hb normal de uno de sus padres y la Hb S del otro progenitor, presentando el rasgo falciforme Hb (AS) y 25 % de probabilidad de que el hijo herede la Hb S de ambos padres Hb (SS) manifestando la enfermedad. (Fig. 2)
DOBLE HETEROCIGOTO HB (SC)
La hemoglobinopatía SC constituye un síndrome doble heterocigótico con patrón de herencia autosómico recesivo, resultado de la herencia del gen S de un padre y del gen C del otro progenitor, se caracteriza por la sustitución del acido glutámico por lisina en la posición 6 de la cadena beta de la hemoglobina.
La hemoglobinopatía SC produce un cuadro clínico menos grave que el de la hemoglobinopatía SS. El crecimiento y el desarrollo sexual son normales, la anemia es leve y las crisis vasoclusivas escasas. Suele palparse esplenomegalia de pequeño tamaño.5,6
HEMOGLOBINA S-BETATALASEMIA: Sβ+tal
Las betatalasemias son el resultado de la falta de síntesis de las cadenas beta de globina. Los genes beta se encuentran en el cromosoma 11, junto con los genes delta y gamma.
Debido al aumento de la frecuencia de los genes, tanto de la Hb S como de la talasemia β en grupos de poblaciones similares, la herencia de ambas anomalías es relativamente frecuente. La combinación Hb S-betatalasemia produce un cuadro clínico de gravedad similar al de la drepanocitosis.5,7
PRESENTACIÓN CLÍNICA
Diferentes tipos de hemoglobina son producidos durante la vida embrionaria, fetal y adulta; cada una de estas, consta de un tetrámero de cadenas polipeptídicas de globina: un par de cadenas alfa (141 aminoácidos), y un par de cadenas beta (146 aminoácidos). La principal hemoglobina que encontramos en el adulto es la Hb A (α2β2), en el feto es la Hb F (α2γ2) y en una pequeña porción del adulto, la Hb A2 (α2δ2). Los eritrocitos aparecen hacia la sexta semana después de la concepción y los tipos principales de hemoglobina son la Portland, Gower I, Gower II. En la semana 10-11, la predominante es la hemoglobina fetal. El cambio a la síntesis de hemoglobina adulta se produce casi exclusivamente a la 38 semanas.9
En los primeros meses de la vida el paciente no presenta síntomas, ya que un elevado porcentaje de su hemoglobina es de tipo fetal, que como se sabe, no contiene cadenas beta. A partir del cuarto o quinto mes casi todas las cadenas gamma de la hemoglobina fetal han sido sustituidas por las cadenas beta patológicas y es a partir de este instante que comienzan a manifestarse los signos y síntomas dependientes del proceso hemolítico. 13
Las manifestaciones clínicas son el resultado de la vaso-oclusión y la hemolisis que conducen a isquemia e infartos tisulares. Es una enfermedad de presentación variable de un individuo a otro, con afectación de múltiples órganos (huesos, pulmones, cerebro, riñón, bazo) y se caracteriza por periodos de crisis repetidas o ausencia de síntomas por tiempo prolongado.
SIGNOS Y SÍNTOMAS
Palidez cutánea, ictericia acentuada con los episodios infecciosos o vasos oclusivos por aumento de la hemólisis. Se presentan cambios esqueléticos por expansión de la médula ósea con deformidades a nivel de la cara y cráneo. El retardo en el desarrollo puberal, así como la hepatomegalia y esplenomegalia son posibles en los primeros años de vida; pues con la edad, el bazo sufre fibrosis progresiva produciéndose auto esplenectomía.8,9
Los pacientes sicklémicos con frecuencia tienen hábitos característicos, extremidades largas, manos y dedos alargados, cráneo en torre y paladar ojival, presentan un tinte ictérico o coloración amarilla de las mucosas como consecuencia de la anemia hemolítica y el depósito de bilirrubina. En la actualidad se describen que todas las manifestaciones clínicas están englobadas en tres crisis fundamentales: 10
1. Crisis vasoclusivasCrisis dolorosas torácicas, dolorosas abdominales y dolorosas articulares con o sin necrosis aséptica, crisis mano - pie, crisis hepáticas, crisis vaso - oclusivas del sistema nervioso central, síndrome torácico agudo.12
Estas crisis se producen por la obstrucción del flujo sanguíneo produciéndose hipoxia (déficit de O2) regional y acidosis lo que aumenta la falciformación y la isquemia. Los factores que pueden precipitarla son la hipoxia, acidosis, deshidratación, infección y la exposición al frío.
2. Crisis de secuestroSe caracteriza por el descenso brusco de la hemoglobina por debajo de 2 g/l, reticulocitosis y secuestro de hematíes en bazo, medula ósea e hígado con una alta mortalidad consecutiva al shock hipovolémico.12
3. Crisis hematológicasSon aquellas en las que existe una disminución brusca de la hemoglobina, con reticulopenia y las más comunes son las aplásticas y la megaloblástica.12
El embarazo en la mujer con Hb SS es más frecuente a medida que aumenta la sobrevida. El pronóstico en relación con la morbilidad y mortalidad materna y fetal ha mejorado ostensiblemente. Sin embargo, todavía constituye una de las situaciones más peligrosas por las que puede atravesar una embarazada.
El cuadro clínico es importante porque si existen manifestaciones severas pre-existentes, el embarazo agrega un factor de riesgo. La morbilidad aumenta del primero al tercer trimestre y es de aproximadamente 80 %.8
Las complicaciones más frecuentes son: exageración de la anemia, crisis vaso-oclusiva dolorosa, síndrome torácico, infección urinaria, preeclampsia, convulsiones, tromboflebitis, sepsis.
Generalmente el dolor es frecuente, presentan cierto grado de disfunción cardiaca por hipertrofia ventricular, hay aumento de la precarga y disminución de la poscarga con fracción de eyección normal y gasto cardiaco alto.8
La anemia falciforme cursa con anoxia tisular; así, la placenta no está exenta de que zonas de tejido se necrosen por secuestro de eritrocitos en la circulación que la perfunde. Esto quedó demostrado en un estudio de Olivar Castejón y cols., donde manifiestan cambios degenerativos del trofoblasto por métodos microscópicos por falta de oxígeno con necrosis tisular, dado que los eritrocitos anormales no aportan la cantidad suficiente de oxigeno; demostrando la reacción de Tenney-Parker, indicativa de la hipoxia crónica y se localiza en regiones de mala perfusión placentaria de origen materno o fetal.
Si el infarto placentario es considerado como una lesión vellositaria degenerativa subsecuente a una oclusión vascular materna y el embarazo con enfermedad drepanocítica transcurre con crisis vaso-oclusivas las consecuencias adversas para el feto son obvias. En gran parte, las manifestaciones fetales son paralelas a las maternas, es decir, que mientras la enfermedad no comprometa la oxigenación materna, el pronóstico fetal es bueno. Sin embargo, se ha encontrado que hay mayor asociación de infertilidad, aborto espontáneo, nacidos muertos, restricción del crecimiento intrauterino y trabajo de parto prematuro en mujeres con hemoglobinopatías falciformes.11
Se debe prestar atención a las crisis dolorosa y su ubicación anatómica dado que pueden simular un abdomen agudo y puedan ser confundidos con embarazo ectópico, apendicitis etc.11,15
Las características principales del dolor drepanocítico crónico incluyen las respuestas emocionales, conductuales, afectivas y psicológicas.
Para las mujeres con anemia falciforme (AF), el embarazo es una situación potencialmente grave que puede dejarla aún más frágil e insegura por la alta incidencia de complicaciones durante la gestación. Ellas conviven con el sentimiento positivo de embarazarse, tener hijos y de la realización traída por la maternidad, miedos y ansiedad marcan la gestación de las mujeres con anemia falciforme. El deseo de ser madre es frustrado por el aborto espontáneo o selectivo, por lo que la atención debe ser diferenciada para que contribuya al mejoramiento no solo físico sino también psíquico de la gestante con drepanocitosis.13,14
Una de las primeras descripciones de complicaciones asociadas al embarazo en pacientes con AD fue realizada por Koback en 1941, quien informó en 37 gestaciones, 33 % de mortalidad materna, 12 abortos espontáneos y 6 nacidos muertos. Desde esa fecha, varios investigadores han publicado sus experiencias sobre morbilidad y mortalidad durante el embarazo.
Los riesgos asociados a las embarazadas y sus hijos con drepanocitosis han disminuido considerablemente durante la década pasada, y se pone como límite el año de 1972. Se realizó una comparación entre lo encontrado antes y después de esta fecha; así, entre 156 embarazos con drepanocitosis, la muerte materna presentó una variación de 4,1 % a 1,7 % respectivamente; los índices de mortalidad fetal y perinatal disminuyeron de 52,7 % a 22,7 %. También se encontró una mejora significativa del peso al nacimiento y un aumento en el número de semanas al final de la gestación de 34,7 semanas a 37,4 semanas.16
Hay circunstancias especiales durante el embarazo que aumentan de modo apreciable la morbilidad de estas mujeres. La bacteriuria asintomática y la pielonefritis aguda están aumentadas sustancialmente, y es importante vigilar cuidadosamente la bacteriuria y erradicarla para prevenir la mayoría de las infecciones urinarias sintomáticas. Si se desarrolla la pielonefritis, estos eritrocitos son extremadamente susceptibles a las endotoxinas, lo cual puede producir una rápida y manifiesta destrucción de los glóbulos rojos mientras se suprime simultáneamente la eritropoyesis.
DIAGNÓSTICO
El diagnostico se realiza mediante la electroforesis de hemoglobina, es un simple análisis de sangre que puede ser realizado a todas las gestantes.16,17
La hemoglobina oscila entre 6 y 8 g/dl.
La anemia normocítica normocrómica, aunque puede ser microcítica cuando se asocia a una talasemia o a un déficit de hierro. Reticulosis por encima de 5 %.
DIAGNÓSTICO PRENATAL
El diagnostico prenatal representa la más importante aplicación de la tecnología en humanos. El programa Nacional para la prevención de Anemia drepanocítica establecido en Cuba en 1983, se basa en la detección de las parejas de riesgo mediante la pesquisa en las mujeres embarazadas, asesoramiento genético y diagnóstico prenatal. Se les brinda la oportunidad de interrumpir el embarazo en los casos que el feto este afectado,18 lo que a su vez permite reducir la incidencia de la enfermedad. El uso del aborto selectivo constituye, para muchos escépticos, un proceder no ético siendo necesario aclarar que las enfermedades moleculares como la Sicklemia, no tienen posibilidad de cura definitiva, por ser un defecto estructural predeterminado genéticamente.
El diagnóstico prenatal (DPN) consiste en la detección de anomalías genéticas o congénitas en el feto. Identificado entre los grandes dilemas en la práctica de la genética médica. Numerosos estudios demuestran diferentes actitudes de las personas en el mundo sobre este tema.
Los lineamientos éticos del DPN han sido recientemente publicados por la OMS incluyen los siguientes:
1. Los servicios del DPN deben estar disponibles de acuerdo a la necesidad médica, independientemente de la capacidad de pago.
2. El asesoramiento genético no directivo debe preceder al DPN e incluir una discusión exhaustiva de sus riesgos, beneficios y limitaciones.
3. El DPN debe ser optativo y voluntario.
4. La pareja debe ser informada objetivamente de todo hallazgo clínico pertinente del DPN.
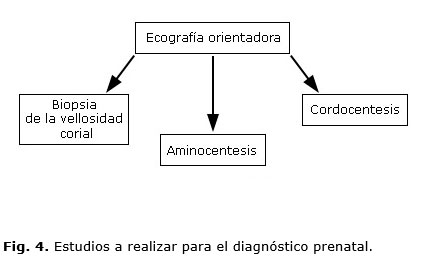
Las técnicas más comunes para obtener material fetal (células y sangre) son las denominadas invasivas, llamadas de esta forma por no respetar el medio interno, ni la integridad de las membranas ovulares, la biopsia de vellosidad (BVC) se realiza entre las semanas 10 y 12 de la gestación, la amniocentesis entre la semana 14 y 20 y la cordocentesis (extracción de sangre fetal de la vena umbilical) a partir de la semana 18 estas son pruebas fiables sin embargo someten al embarazo a un riesgo no despreciable de perdida fetal debido al propio proceder.
Todas estas técnicas invasivas (BVC y amniocentesis) presentan los mismos riesgos: rotura de la bolsa amniótica, desencadenamiento del parto, metrorragias-hemorragias anormales (10-50 %), punción del cordón umbilical y la subsiguiente hemorragia, infecciones (0,3 %), hematomas retrocoriales o intracoriales, punción de órganos vitales del feto (corazón o cerebro) y abortos, relacionados con la experiencia del operador, número de punciones necesarias y la vía utilizada.19,20
La aceptación o el rechazo de un individuo, un grupo o una sociedad hacia el diagnóstico prenatal y la interrupción del embarazo de fetos afectados están determinados por las respuestas a las siguientes preguntas:
- ¿En qué momento de la gestación se considera al producto de la concepción como una persona con derechos?
- ¿Cuál es la importancia relativa de los derechos del feto comparados con los de la madre?
- ¿Es la vida (cualquiera que sea su calidad) mejor que la ausencia de la vida?
Por supuesto, no existe ninguna garantía de que ambos miembros de la pareja respondan de igual forma a estas preguntas, sino que pueden hacerlo de forma muy diferente.18
Cuando el feto resulta afectado, de inmediato surgen problemas con estas definiciones.
¿Es aborto interrumpir el embarazo en el estado de embrión? Según la lengua castellana sí, según la embriología no; en clínica se hablaría de un aborto: ¿es aborto interrumpir el embarazo cuando el feto no es viable ni dentro ni fuera del útero?18 ¿Es el embrión o el feto un ser humano en el momento del aborto?
Las respuestas a las interrogantes de cuándo el feto se considera persona? o ¿cuándo el feto se considera paciente, se encuentran entre las más controvertidas de la medicina moderna y constituyen grandes dilemas éticos relacionados con el comienzo de la vida. Algunos consideran que desde etapas precoces de la gestación el embrión y el feto tienen un estatus moral y por lo tanto debe ser respetado como una persona potencial. El reconocimiento del estatus moral del embrión y de sus derechos como persona se encuentra en el centro de los grandes conflictos éticos actuales.18
De forma general podemos decir que el estatus moral del feto está muy relacionado con la decisión de los padres de aceptar o no al feto enfermo, pues este no tiene capacidad para decidir. Resulta muy importante que el médico a pesar de sus convicciones morales, no trate de influir de ninguna manera en la decisión paterna.
No son pocos los que acusan al diagnóstico prenatal de ser una práctica eugenésica, porque se aborta el feto enfermo y se acepta el feto sano. Por eso, algunos sectores acuden al concepto de "calidad de vida" para tratar de justificar o no el aborto selectivo.18,20
TRATAMIENTO
Aunque no hay ninguna cura para la anemia falciforme los médicos pueden prestar considerable ayuda a quienes padecen la enfermedad, por lo que tendremos varios tipos de tratamiento:
1. Preventivo
2. Específico de la enfermedad
El tratamiento preventivo está basado en el diagnostico prenatal que se explicó anteriormente para evitar el nacimiento del feto afectado de la enfermedad.
No hay datos sistemáticos que puedan ser usados como guía ponderal de manejo de crisis dolorosa aguda en la paciente embarazada.22
En la CVO dolorosa la medida terapéutica más importante es la hidratación por vía oral o parenteral, según la intensidad, la localización y las manifestaciones que acompañan a la crisis (dolor abdominal, vómitos) se utilizan soluciones de dextrosa. Los analgésicos más utilizados en nuestro país son la dipirona y el espasmoforte. El uso de narcóticos en estos pacientes no es aconsejable.
La Hidroxiurea disminuye fenómenos vaso-oclusivo en pacientes con enfermedad de células falciformes pero tiene acción teratogénica durante el embarazo (Fig. 4).
INDICACIONES DE LA TRANSFUSIÓN
Se utilizan en la crisis aplástica y de secuestro, en las infecciones severas sobre todo pulmonares y en la insuficiencia cardiaca, pulmonar o renal crónicas.
En la Hb SS se indican las transfusiones:21,22
1. Para mejorar la capacidad de transporte de oxígeno de la sangre.
2. Anemia severa que produzca disnea, hipotensión postural, angina, insuficiencia cardiaca, disfunción cerebral.
3. Hipoxia aguda o crónica (PO2 ≤ 70 torr).
4. Para mejorar la perfusión en la microcirculación y disminuir la proporción de hematíes con HBS.
5. Infecciones graves.
6. Crisis vasooclusivas del sistema nervioso central (SNC).
7. Crisis de secuestro esplénica o hepática.
8. Síndrome torácico agudo.
La exanguíneo-transfusión se aplica cuando es necesario disminuir de manera rápida el porcentaje de Hb S a menos de 30,0 - 40,0 %. Es un método rápido se extrae la sangre de una vena al mismo tiempo que se transfunde por otra. Se recambian 500 mL y el total a recambiar es de 3,0 - 4,0 L. Si con una exanguíneo -transfusión no se logra el porcentaje de Hb S deseado se repite al día siguiente.
Toda la sangre será estudiada con la prueba de solubilidad para descartar el donante AS y con las pruebas para el diagnóstico de sífilis, hepatitis B y C y VIH-SIDA.
A toda paciente se le hará el fenotipo de sus hematíes: ABO, Rh, Kell, Duffy, Kidd .
En la sangre del donante los más importantes son: Sistema Rh, Kell, Duffy
El uso profiláctico de la transfusión o exanguíneo -transfusión se discute.25 Los que apoyan este tratamiento aducen que al disminuir el porcentaje de Hb S disminuyen los fenómenos vaso-oclusivos en la placenta, las complicaciones y la morbilidad y mortalidad materna y fetal; otros no encuentran diferencias. Por otro lado, 22 % de las mujeres transfundidas desarrollan alo-anticuerpos anti-eritrocitarios, algunos de los cuales pueden causar anemia hemolítica del recién nacido.
En lo que se refiere a los métodos contraceptivos también existen controversias. Se describen complicaciones con la utilización de contraceptivos orales, progesterona de depósito y dispositivos intrauterinos; por lo que se aboga por la esterilización quirúrgica siempre que sea posible con paridad satisfecha y signos de enfermedad descompensada.
Se concluye que es menester un alto índice de perspicacia y buen diagnóstico para obtener resultados óptimos en las pacientes embarazadas afectadas, de enfermedad de células falciformes. Por lo que el seguimiento será multidisciplinario con obstetra, hematólogo, intensivista y anestesiólogo.
Conflicto de interéses
Los autores no delcaran conflictos de interéses.
REFERENCIAS BIBLIOGRÁFICAS
1. Rogers DT, Molokie R. Sickle Cell Disease in Pregnancy. Obstet Gynecol Clin North Am. 2010;37(2):223-37.
2. Ferreira A, Henri A, Marguti I, Bechmann I, Beuzard Y, Jeney V, et al. Sickle hemoglobin confers tolerance to Plasmodium infection. Cell. 2011; 145:398-409.
3. Peñaloza RI, Buentello L, Hernández MA, Nieva B, Lisker R, Salamanca F. Frecuencia de la hemoglobina S en cinco poblaciones mexicanas y su importancia en la salud pública. Salud pública Méx. 2008;50(4).
4. Svarch E. Fisiopatología de la drepanocitosis. Rev Cubana Hematol Inmunol Hemoter. 2009;25(1).
5. Kato G, Hebbel R, Streinberg M, Gladwin M. Vasculopathy in Sickle Cell Disease: biology, pathophisiology, genetics, translational medicine and new research direction. Am J Hematol. 2009:618-25.
6. Al Jama FE, Gasem T, Burshaid S, Rahman J, Al Suleiman SA, Rahman MS. Pregnancy outcome in patients with homozygous sickle cell disease in a university hospital. Eastern Saudi Arabia. Arch Gynecol Obstet. 2009;280:793-7.
7. Belisário AR, Martins ML, Brito AM, Rodrigues CV, Silva CM, Viana MB. Globin gene cluster haplotypes in a cohort of 221 children with sickle cell anemia or S_0-thalassemia and their association with clinical and hematological features. Acta Haematol. 2010;124:162-70.
8. Xavier ASG. Experiências reprodutivas de mulheres com anemia falciforme. Salvador. Dissertação [Mestrado em Enfermagem]- Universidade Federal da Bahia; 2011.
9. Booth C, Inusa B, Obaro S.Iinfection in Sickle Cell Disease. Internat J Infect Dis. 2010;14:2-12.
10. Nomura RM, Igai AM, Tosta K, Fonseca GH, Gualandro SF, Zugaib M. Resultados maternos e perinatais em gestac¸ões complicadas por doenc¸as falciformes. Rev Bras Ginecol Obstet. 2010;32:405-11.
11. Al-Farsi SH, Al-Riyami NM, Al-Khabori MK, Al-Hunaini MN. Maternal complications and the association with variables in pregnant women with sickle cell disease. Hemoglobin. 2013;37:219-26.
12. Parrish MR, Morrison JC. Sickle cell crisis and pregnancy. Semin Perinatol. 2013;37:274-9.
13. Martín MR, Duany E, Domínguez M, Alfonso K, Santana ME, Viñales MI. Anemia Falciforme: Conocimientos y percepción actual del riesgo en jóvenes detectados al nacimiento como portadores sanos. Rev Cubana Genet Comunit. 2008;2(3).
14. Santos NS, Chvatal VLS, Varga CRR, Böttcher-Luiz F, Turato ER. Vivências sobre gravidez relatadas por mulheres com anemia falciforme em hospital universitário: um estudo qualitativo. Revista Psicologia e Saúde. 2011;3(2):23-9
15. Monken FV, Barros NN, Valadares PJC, Macedo RSPB, Cruz SG, Cury PS, et al. Situações de urgência na gestante com doença falciforme. Rev méd Minas Gerais. 2010;20(S2):73-7.
16. Martín MR, Lemus MT, Marcheco B. El programa cubano de prevención de Anemia Falciforme. Resultados del periodo 1990-2005. Rev Cubana Genet Comunit. 2008;2(2):59-66.
17. Fernández J, Pérez A, Fragoso M, Rivero R. El diagnóstico temprano de la anemia falciforme: un problema no resuelto en África negra. Rev Cubana Hematol Inmunol Hemoter. 2012;28(2):195-7.
18. González Y, Martín MR. Cumplimiento de principios éticos y normativos en la indicación de la electroforesis de hemoglobina a gestantes. Rev Panorama Cuba y Salud. enero-abril. 2009;4(1):44-53.
19. Taboada N, Gómez M, Algora AE, Noa ME, Arcas G, Noche G, Herrera M. Resultados del Programa de prevención de hemoglobinopatías SS y SC en el periodo 1987-2007 en la provincia Villa Clara, Cuba. Rev Cubana Genet Comunit. 2010;4(1):605-14.
20. Estrada M, Machado G, Estenoz Y. Situación actual del programa de prevención prenatal de sicklemia en el municipio Ciego de Ávila durante 2011. MEDICIEGO. 2012;18(S1)
21. Ngo^ C, Kayem G, Habibi A, Benachi A, Goffinet F, Galacte´ros F, et al. Pregnancy in sickle cell disease: maternal and fetal outcomes in a population receiving prophylactic partial exchange transfusions. Eur J Obstet Gynecol Reprod Biol. 2010;152:138-42.
22. Svarch E. Programa cubano de atención integral al paciente con drepanocitosis. Rev Cubana Hematol Inmunol Hemoter. 2011;27(2):165-7.
23. Royal College of Obstetricians and Gynaecologists Green-top, Guideline. Management of sickle cell disease in pregnancy. RCOG. 2011;61:1-20.
24. Ngo C, Kayem G, A Habibi, Benachi A, Goffinet F, Galactéros E, et al. Embarazo en la enfermedad de células falciformes: resultados maternos y fetales en una población que recibe exanguinotransfusión parcial profilácticos. Eur J Obstet Gynecol Reproductive Biology. 2010.
25. Hernández C, Téllez ML, Espinosa E. Impacto social de la atención integral del embarazo en la drepanocitosis. Scientific Meeting of the International Day of sickle cell disease (sickle cell), saluting the 150th anniversary of the Cuba Academy of Sciences and the International Year of African descent. 2011.
Recibido: 29 octubre 2015.
Aprobado: 30 enero 2016.

