










 Servicios personalizados
Servicios personalizados Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkMalaria is still one of the most serious parasitic diseases affecting humankind, causing almost half million deaths annually.1 Parasites from the genus Plasmodium are the causative agents of the disease, among which P. falciparum causes great morbidity and mortality. The Plasmodium life cycle is divided into two phases: sporogonic (within the mosquito host) and schizogonic (within the mammal host).2 The erythrocytic cycle occurs during the schizogonic phase, causing the symptoms of the disease (cyclic fevers, headache and trembles), due to the infected cell rupture and the release of new parasites. During the erythrocytic cycle, the parasite develops through four stages: merozoite, ring, trophozoite and schizont.2
Proteases can initiate and/or regulate essential biological processes such as apoptosis, cell cycle control and cell migration. Given the significance of proteolytic events for many organisms, it is expected that the same occurs in Toxoplasma and Plasmodium apicomplexan parasites.3 During the intraerythrocytic stages of P. falciparum there is a broad range of temporal expression for various classes of proteases.4 Decades of parasite biology research has allowed defining the role of proteases in essential physiological processes such as hemoglobin degradation, erythrocyte invasion and egress.3
The already proved role of proteolytic events on the life cycle of the parasite led to the study of its proteases individually and the search for new inhibitors of the natural/recombinant enzymes (reviewed by.3,5 However, considering that proteolytic events usually involve more than one enzyme, and that they are differentially expressed during the cell cycle, the study of catalytic activity as a whole arises as an alternative for a better understanding of the parasite´s biology and potential targets for compromise the parasite development.
In parasites, only a few general proteolysis studies had been conducted. As a source of proteases, cell lysates were used for the studies in P. falciparum6 and Leishmania (L.) amazonensis.7 However, the use of isolated living cells, which allow to measure proteolysis in physiological conditions, was mainly reported for Plasmodium asynchronous cultures.8,9,10 For example, the analysis of calcium effects on Fluorescence Resonance Energy Transfer (FRET) substrates8 and the development of a specific calpain activity assay with the substrate Z-Phe-Arg-MCA.9 Moreover, the use of isolated parasites from asynchronous cultures was also reported in experiments to correlate inhibition of proteolysis with impairment of P. falciparum growth in vitro by potential antimalarial drugs. In this context, our group described two class of compounds: i) organotellurium compounds inhibit Z-Phe-Arg-MCA proteolysis in living cells and P. falciparum growth in vitro10 and ii) bestatin-derived peptidomimetics inhibit P. falciparum growth in vitro, PfAM1 activity in vitro and also Ala-MCA proteolysis in isolated living cells.11
However, to better understanding the proteolytic events during parasite´s life cycle as well as to correlate inhibition of proteolysis at specific asexual blood stage with impairment of P. falciparum growth in vitro it is required to extend the hydrolytic studies in synchronized P. falciparum cultures at all asexual blood stages. The class of cysteine proteases is the most representative within the total protease activity of the parasite. P. falciparum genome analysis identified 92 possible genes for proteases, 33 of which correspond to cysteine proteases.12 As these proteases are involved in essential processes for parasite such as the egress and invasion of parasites in erythrocytes or hepatocytes and also the acquisition of amino acids from the degradation of hemoglobin.13 The development of inhibitors for these enzymes have a relevant antimalarial potential,14 although is necessary a selective quantification of proteolysis during the asexual blood stages of P. falciparum isolated parasites and hence to develop new tools for drug discovery, which is the aim of the present work.
In order to accomplish this aim P. falciparum 3D7 strain15 was cultured in RPMI 1640 (Gibco) supplemented with Albumax II (Gibco) 0.5 %, employing a method based on.16 The parasites were synchronized with sorbitol using a procedure described elsewhere.17 To collect the parasites, synchronized cultures were cultivated and parasite maturation was followed through Giemsa-stained smears. Cultures containing rings, trophozoites, early schizonts or late schizonts/merozoites were used for parasite collection. Parasites were isolated as previously described,18 maintained in MOPS buffer (3-(N-morpholino) propanesulfonic acid; 50 mM; NaCl 116 mM; KCl 5,4 mM; MgSO4·7H2O 0.8 mM; glucose 5.5 mM and CaCl2 2 mM, pH 7.2) on ice during the experiments (no more than three hours) and parasite´s integrity was verified through Giemsa-stained smears after finishing the assays. A sample of parasite´s suspension was always kept at -20 oC and afterwards the lysates were obtained by BugBuster® Master Mix (Novagen, EUA) treatment, according to fabricant instructions. The protein concentration of parasite´s suspension was measured by Bradford's method using BSA (bovin serum albumin from Sigma) as standard.19
The fluorogenic substrate Z-Phe-Arg-MCA was chosen in our studies because it is a broad specificity substrate (serine and cysteine proteases) and has been used previously in parasite living cells proteolysis assays.9,10
The hydrolysis of Z-Phe-Arg-MCA (λex= 380 nm and λem= 460 nm, slit parameter of 10/10 nm) was monitored for 10 min in a spectrofluorometer Synergy (BioTek Instruments Inc., USA), at 37 °C. Isolated parasites were transferred to a 96-well plate (enough quantity to reach a slope of the curve Fluorescence vs. time of at least20 containing MOPS buffer (final assay volume 200 µL). The assay was initiated by addition of 10 μM of the substrate. To enable the quantification of released MCA a calibration curve was developed (Fluorescence vs. degraded substrate) incubating increasing quantities of Z-Phe-Arg-MCA with isolated parasites as source of enzymes. One unit of enzymatic activity was defined as the amount of enzyme able to release 1 µmol of MCA per min under specified conditions(cotcal curve= 0.000126658 µmol·L-1/F). The specific activity was expressed as enzymatic activity units related to protein concentration in the assay.
In inhibition assays, the parasites were pre-incubated 30 min at 37 oC with inhibitors for the different class of proteases: serine proteases (phenylmethylsulfonyl fluoride (PMSF) (0.5 mM); aspartyl proteases, pepstatin A (1 µM); metalloproteases, 1,10-phenanthroline (5 mM), and cysteine proteases, trans-epoxysuccinyl-L-leucylamido-(4-guanidine) butane (E64) (5 µM). After incubation, the assays were carried out in the conditions described above. As control, parasites were incubated in the same conditions without the inhibitors. Reducing agents promote a proteolysis increase in non-covalent inhibited cysteine proteases, which reveals remaining active proteases. Thus, to determine inhibition reversibility by reducing agents as dithiotreitol (DTT) each experiment was performed with a further addition of DTT 5 mM for 10 min measurement. All results are expressed as mean ± SD of three individual experiments (with three replicates each one). ANOVA was used for comparisons of more than three groups, followed by Tukey or Dunnet post-tests. A p value less than 0,05 was considered indicative of a statistically significant difference. GraphPad Prism software (San Diego, CA, USA) was used for all statistical tests.
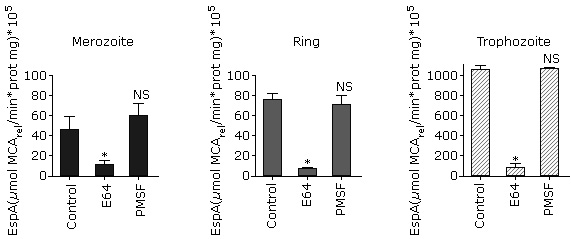
As shown in figure 1, A, a higher specific activity was observed in trophozoites and early schizonts. This result is similar to that obtained for parasite´s lysate proteolytic activity using FRET substrates.6 The substrate Z-Phe-Arg-MCA cannot be degraded by exopeptidases because it has both its C- and N-terminal portions blocked so that only endopeptidases can hydrolyze it. In P. falciparum, this substrate can be degraded by cysteine proteases such as falcipains (FP-1, FP-2, FP-2´ and FP-3)20 and calpain.9
 A: Comparison of Z-Phe-Arg-MCA proteolysis in isolated P. falciparum parasites at the four erythrocytic stages. The data were compared with one way ANOVA followed by Tukey pos-test. B: Effect of typical protease inhibitors on Z-Phe-Arg-MCA proteolysis at P. falciparum asexual blood stages.The data were compared with one way ANOVA followed by Dunnett post test. In all instances, asterisks indicate significant differences with p< 0.05 for the comparison between groups (A) or the comparison with the control (B) (n= 3, three independent experiments).
A: Comparison of Z-Phe-Arg-MCA proteolysis in isolated P. falciparum parasites at the four erythrocytic stages. The data were compared with one way ANOVA followed by Tukey pos-test. B: Effect of typical protease inhibitors on Z-Phe-Arg-MCA proteolysis at P. falciparum asexual blood stages.The data were compared with one way ANOVA followed by Dunnett post test. In all instances, asterisks indicate significant differences with p< 0.05 for the comparison between groups (A) or the comparison with the control (B) (n= 3, three independent experiments).Fig. 1 Profile of Z-Phe-Arg-MCA hydrolysis by isolated P. falciparum parasites at asexual blood stages.
Next, in order to determinate the class of proteases that are acting on this substrate, classical protease inhibitors were tested (Fig. 1, B). There is no significant inhibition observed by pepstatin A or 1.10-phenanthroline, which considering that aspartic and metalloproteases, respectively, are expressed during the intraerythrocytic stages4,21 indicates that Z-Phe-Arg-MCA is not degraded by these enzymes at any erythrocytic stage. This outcome is expected, taking into account the specificity described for P. falciparum metalloendopeptidases such as falcilisin,22 metalloaminopeptidases (reviewed in23) and likewise for aspartic proteases such as plasmepsins.24 Similar results were obtained in trophozoites by Gomes and coworkers using similar conditions. 9
On the other hand, E64 inhibits the proteolytic activity in all stages tested, suggesting that cysteine protease activity is present (Fig. 1, B). These results are in accordance with those previously obtained in similar conditions for trophozoites9 and for recombinant falcipain 2 and 3.25
In addition, there is a significant decrease of proteolytic activity in the presence of PMSF in merozoites, rings and trophozoites whereas there is no inhibition in schizonts (Fig 1, B). In the case of trophozoites, these results differ with that obtained by Gomes et al. (2014), which did not observe PMSF inhibition with this substrate. These differences could be due to the cell-inhibitor incubation time and the inhibitor concentration. In the present work, we incubated 0.5 mM of inhibitor for 30 min instead of 0.01 mM for 10 min used in that study,9 which probably allowed a better enzyme-inhibitor interaction.
Nevertheless, taking into account that PMSF is a serine protease inhibitor but it could also inhibit cysteine proteases and that inhibition can be reversed by DTT,26 a reversibility inhibition assay was performed. As shown in figure 2, DTT addition annulled PMSF inhibition but had no effect in cysteine protease inhibition by E64. This result indicates that the PMSF inhibition observed was the result of unspecific interaction with cysteine proteases and therefore the serine protease activity is very low or null under these conditions.
 The data were compared with one way ANOVA followed by Dunnett post test. Asterisks indicate significant difference with p< 0.05 compared to control. (n= 3, three independent experiments).
The data were compared with one way ANOVA followed by Dunnett post test. Asterisks indicate significant difference with p< 0.05 compared to control. (n= 3, three independent experiments).Fig. 2 Effect of PMSF reversible inhibition of Z-Phe-Arg-MCA proteolysis in isolated P. falciparum parasites.
The results suggested that the main enzymatic activity observed in these conditions is due to cysteine proteases in accordance with previous studies using isolated trophozoites with the same substrate9 or in parasite lysates with broad specificity FRET peptides.6 Considering the present work and the differential expression of proteases during P. falciparum erythrocytic cycle,4,21,25,27,28 it can be postulated that the proteases responsible for the hydrolysis of Z-Phe-Arg-MCA are mostly cysteine proteases in merozoites (FP-1), rings (FP-1 and Pfcalpain), trophozoites (FP-2, 2´, 3 and Pfcalpain) and schizonts (FP-3, SERA6 and Pfcalpain). Our results shows a interestingly proteolytic profile of cysteine proteases during the asexual blood stages and the most active period observed is trophozoite stage, which is 20 fold higher than schizont and 40 fold against ring/merozoite.
Taken together, this is the first study of proteolysis in parasite living cells isolated from synchronous cultures of P. falciparum asexual erythrocytic stages, and it establishes a routine assay that may be useful: i) to identify new inhibitors effect on endogenous stage specific proteolysis and indication of suitable therapeutic window and ii) to determine the mechanism of action for molecules with antimalarial activity in vitro.

