










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El 11 de marzo de 2020, la Organización Mundial de la Salud (OMS) declaró la pandemia de COVID-19 originándose una emergencia de salud pública sin precedentes. El agente causal del brote reportado inicialmente en Wuhan, China en diciembre de 2019 fue un nuevo tipo de virus de la familia Coronaviridae, que posteriormente fue denominado como SARS-CoV-2.1,2
Los coronavirus pertenecen a una familia de virus que causan infección en los seres humanos y en una variedad de animales. Los coronavirus que afectan al ser humano (HCoV-NL63, HCoV-229E, HCoV-OC43 y HKU1) pueden producir cuadros clínicos que van desde el resfriado común con patrón estacional en invierno hasta otros más graves como los producidos por los virus del Síndrome Respiratorio Agudo Grave (por sus siglas en inglés, SARS) y del Síndrome Respiratorio de Oriente Próximo (MERS-CoV). El SARS-CoV-2 constituye el séptimo coronavirus capaz de provocar infecciones en humanos.2
El diagnóstico virológico se realiza mediante la detección del ARN viral a través de la técnica de PCR en tiempo real (PCR-TR), es la técnica más fiable y ampliamente utilizada por lo que se considera el estándar de oro del diagnóstico. Esta técnica permite detectar la presencia del virus en las etapas iniciales de la infección, incluso antes de la aparición de los síntomas. También existen los ensayos de detección de antígeno, las cuales se basan en la detección de proteínas virales específicas de SARS-CoV-2 en la muestra, como la proteína N y las subunidades S1 o S2 de la proteína espiga. La detección del antígeno viral implica replicación del virus por lo que un resultado positivo de la prueba indicaría infección actual por SARS-CoV-2.3,4,5
Existen diversos métodos de detección de antígenos basadas en inmunocromatografía (pruebas rápidas) que ofrecen ventajas como la rapidez y sencillez y no requieren infraestructura especializada. Otras, están basadas en inmunofluorescencia, técnicas inmunoenzimáticas o de electroquimioluminiscencia que precisan de instrumental y personal de laboratorio especializado. Estas técnicas ofrecen ventajas en términos de tiempos de respuesta más cortos y costos reducidos, especialmente en situaciones en las que la capacidad de la prueba de PCR-TR es limitada.6,7,8
En el contexto de la pandemia de COVID-19, Cuba se une al esfuerzo mundial de lograr técnicas con buen desempeño analítico y factura nacional. El Laboratorio de Virología del IPK se ha encargado de la evaluación de los mismos, y ya se encuentran en fase de aplicación los ensayos de detección de anticuerpos.9 El ensayo UMELISA SARS-CoV-2 Antígeno es un ensayo desarrollado para la detección de antígenos correspondientes a la proteína de la nucleocápside viral (N) en muestras de exudado nasofaríngeo procedentes de individuos con sospecha de COVID-19. En el presente artículo se describe la evaluación clínica del mismo como paso previo a su introducción en el diagnóstico y la vigilancia de COVID-19.
Métodos
Para medir la especificidad y sensibilidad del UMELISA SARS-CoV-2 Antígeno, se utilizó un panel de muestras clínicas positivas y negativas procedentes de la vigilancia y el diagnóstico y analizadas previamente por PCR-TR a SARS-CoV-2. Se seleccionaron muestras colectadas en medio de transporte sin guanidium, SDS, entre otras, según instrucciones del fabricante. Se escogieron muestras colectadas con 24 y 48 horas, cuyo medio de transporte no presentara turbidez o con cambio de color aparente y que fueran transportadas en medio de transporte viral BTV, BIOCEN, Cuba y en medio de transporte CITOSWAB VTM, China. A continuación, se describen dichos paneles:
Muestras clínicas
Panel de muestras de exudado nasofaríngeo negativas al SARS-CoV-2 para estudio de especificidad clínica. Se utilizaron 123 muestras (73 con 48 hrs de colectadas y 50 con 24 hrs de colectadas).
Panel de muestras de exudado nasofaríngeo positivas a diferentes agentes virales infecciosos que producen infecciones respiratorias agudas (IRA) para estudio de especificidad analítica. Se utilizaron 8 muestras, caracterizadas de la siguiente forma: positivas a Influenza A (n=5), Parainfluenzavirus 3 (n=1), coronavirus humanos estacional OC43 (n=1) y Rinovirus (n=1).
Panel de muestras de exudado nasofaríngeo positivas al SARS CoV-2 (n=121). Según las características clínicas, epidemiológicas y de laboratorio a que pertenecían estas muestras se utilizaron: muestras para confirmar procedentes de otros centros de diagnóstico molecular del SARS CoV-2 n=41 (33,8 %), contactos n=41 (33,8 %), evolutivos (sin definir días de evolución) n=4 (3,3 %), sospechosos de una IRA n= 9 (7,4 %) y vigilancia n=26 (21,4 %).
Muestras de exudado nasofaríngeo procedentes de casos evolutivos
Se incluyeron además, 27 muestras con resultado positivo por PCR-TR, 17 de casos evolutivos colectadas al 5to. día del primer PCR-TR positivo y 10 del 7mo. día. Las muestras positivas incluidas en este estudio tenían un rango de Ct (umbral de ciclo, del inglés cycle threshold) entre 19 y 38.
Muestras para el Control de calidad del UMELISA SARS-CoV-2 Antígeno
Muestras positivas por UMELISA SARS-CoV-2 Antígeno (n=78) procedentes de dos laboratorios SUMA de La Habana, que fueron analizados por PCR-TR en el IPK.
Extracción de ácidos nucleicos
El proceso de extracción del ARN se realizó de forma automática a partir de una alícuota de 140µL de las muestras clínicas, empleando el extractor automático QIAcube TM (QIAGEN, EUA). Para realizar este procedimiento se utilizaron los materiales y reactivos contenidos en los estuches comerciales QIAamp Viral RNA Mini Kit (QIAGEN, EUA). La preparación de los reactivos se realizó siguiendo las instrucciones del fabricante.
Estuches de PCR-TR
Las muestras de los paneles de muestras negativos y positivos y para el Control de calidad del UMELISA SARS-CoV-2 Antígeno fueron analizadas por el ensayo de PCR en TR STAT-NAT COVID-19 MULTI de la casa comercial SENTINEL DIAGNOSTICS (Milán, Italia). Este ensayo amplifica simultáneamente dos genes del SARS-CoV-2: RdRP y Orf1b. Las muestras positivas incluidas en este estudio tenían un rango de Ct entre 6 y 25. Las muestras correspondientes a casos evolutivos fueron analizados por el ensayo RIDA®GENE SARS-CoV-2 (R-Biopharm, Alemania) que amplifica el gen de envoltura (gen E) del SARS-CoV-2. Las muestras incluidas en este estudio tenían un rango de Ct entre 19 y 38.
UMELISA SARS-CoV-2 Antígeno
El UMELISA SARS-CoV-2 Antígeno es un ensayo inmunoenzimático heterogéneo tipo sándwich de doble anticuerpo para la detección de antígenos correspondientes a la proteína de la nucleocápside viral (N) en muestras de exudado nasofaríngeo procedentes de individuos con sospecha de COVID-19. El ensayo emplea las ventajas de la reacción de alta afinidad entre la Estreptavidina y la Biotina. Este ensayo utiliza como fase sólida, placas de ultramicroELISA revestidas con anticuerpos específicos a la proteína N del SARS-CoV-2. Brevemente, una vez que las muestras son colectadas en medios de transporte viral universales que no contengan sustancias inactivantes, estas requieren de un paso de preparación en el cual la proteína N es extraída, en caso de encontrarse presente. Posteriormente, se incuban en los pocillos de las tiras y los anticuerpos en su superficie capturan los antígenos correspondientes a la proteína N. A continuación, previo lavado que elimina los componentes de la muestra no fijados, se añaden anticuerpos biotinilados específicos a la proteína N, que se unirán al complejo anticuerpo-antígeno formado sobre la fase sólida. Un nuevo lavado eliminará los anticuerpos biotinilados que no reaccionaron y quedaron en exceso. Seguidamente se añade el conjugado Estreptavidina/Fosfatasa Alcalina, que se unirá a las moléculas de biotina y luego de otro paso de incubación y lavado, se adiciona el sustrato fluorigénico (4 Metilumbeliferil fosfato), que será hidrolizado y la intensidad de la fluorescencia emitida permitirá detectar la presencia de antígenos de SARS-CoV-2 en la muestra.10
Condiciones de bioseguridad
El ensayo UMELISA SARS-CoV-2 Antígeno se realizó en condiciones de Bioseguridad Nivel II, utilizando cabina de seguridad biológica Clase II certificada para realizar el paso de dilución de las muestras en placas de 96 pocillos, agitación, adición de las muestras en las placas de reacción y los lavados de las placas. El personal encargado de la realización del ensayo utilizó Equipo de Protección Personal (EPP) completo.
Análisis estadístico
Para realizar la evaluación del ensayo UMELISA SARS-CoV-2 Antígeno se tuvo en cuenta la Regulación No. 47-2007 Requisitos para la evaluación del desempeño de los diagnosticadores del CECMED. Se calculó la sensibilidad clínica, especificidad clínica, especificidad analítica (Reactividad cruzada), prueba de concordancia, valores predictivos positivo (VPP) y negativo (VPN) e Índice Kappa (K). Para el cálculo de estos parámetros se usaron las tablas de contingencia 2 x 2. Las muestras fueron estratificadas según criterios clínico-epidemiológicos y de laboratorio (casos sospechosos, evolutivos y muestras con Ct<30). Para el cálculo de los parámetros se utilizó la aplicación LabCal V1.0.1.11 A todos estos parámetros se les calculó el intervalo de confianza al 95 % (IC 95 %). Para la comparación de la media del cálculo de las fluorescencias (2 valores en el caso de las muestras negativas y 4 en el caso de las muestras positivas) entre muestras positivas y negativas en el ensayo UMELISA SARS-CoV-2 antígeno se utilizó el programa GraphPad Prism V5.0 y la prueba de Mann Whitney, en la que se consideraron significativos los valores del estadígrafo a una p<0.05.
Consideraciones éticas
Se utilizaron muestras clínicas del laboratorio de Virus Respiratorios del Dpto. de Virología, del IPK. Dichas muestras encontraban debidamente almacenadas y organizadas y provienen de las muestras del diagnóstico, la vigilancia y las investigaciones. Todos los participantes tuvieron el compromiso de salvaguardar la confidencialidad de los individuos y de los pacientes positivos al SARS CoV-2 cuyas muestras estarán incluidas en la evaluación. El estudio se realizó siguiendo las Buenas Prácticas Clínicas y de Laboratorio (BPC/BPL) y siguiendo las instrucciones que el fabricante propone, el personal que realizo el estudio estaba calificado y se emplearan métodos para la realización de las pruebas de laboratorio y estadísticas reconocidos a nivel internacional.
Resultados
Especificidad
Al analizar la especificidad clínica y la especificidad analítica o reactividad cruzada de la técnica UMELISA SARS-CoV-2 antígeno utilizando el panel de muestras negativas al SARS CoV-2 y positivas a otros agentes virales obtuvimos los resultados mostrados en la tabla 1.
Tabla 1 Especificidad clínica y analítica del ensayo UMELISA SARS-CoV-2 antígeno
| Muestras | N | Positivo | Negativo | Especificidad % (IC 95 %) |
|---|---|---|---|---|
| Muestras negativas SARS-CoV-2 (48 hrs) | 73 | 2 | 71 | 97,2 (89,5-99,5) |
| Muestras negativas SARS-CoV-2 (24 hrs) | 50 | 0 | 50 | 100,0 (91,1-99,8) |
| Muestras positivas a varios agentes virales que producen IRA | 8 | 0 | 8 | 100,0 (59,7-98,8) |
| Total | 131 | 2 | 129 | 98,4 (94,0-99,7) |
IC 95 %: Intervalos de Confianza del 95 %
Sensibilidad clínica, VPP, VPN, Concordancia e Índice Kappa
Al analizar la sensibilidad de la técnica UMELISA SARS-CoV-2 antígeno utilizando las muestras positivas provenientes de pacientes confirmados al SARS CoV-2 por PCR-TR, obtuvimos que 98/121 resultaron positivos, para una sensibilidad de 80,9 %. Al realizar el análisis con las muestras con un Ct ≤ 33 se incrementó la sensibilidad a 85,5 %. En la tabla 2 se muestran estos resultados con sus IC al 95 % y otros parámetros de la evaluación.
Tabla 2 Sensibilidad clínica, VPP, VPN, Concordancia e Índice Kappa del ensayo UMELISA SARS-CoV-2 antígeno en el total de muestras estudiadas y en aquellas con Ct≤33.
| Muestras | Sensibilidad % (IC 95 %) | VPP % (IC 95 %) | VPN % (IC 95 %) | Concordancia % (IC 95 %) | Kappa (IC 95 %) |
|---|---|---|---|---|---|
| Total de muestras (n=121) | 80,9 (72,6-87,3) | 98,0 (92,2-99,6) | 84,8 (77,9-89,9) | 90,08 (85,5-93,3) | 0,800 (0,726-0,874) (buena concordancia) |
| Muestras con Ct≤33 (n=111) | 85,5 (77,3-91,2) | 97,9 (92,03-99,6) | 88.9 (82,4-93,3) | 92,5 (88,3-95,4) | 0,85 (0,78-0,92) (muy buena concordancia) |
IC 95 %: Intervalos de Confianza del 95 %; VPP: Valor Predictivo Positivo; VPN: Valor Predictivo Negativo; Ct: umbral de ciclo
En la Figura 1 se muestra el análisis del ensayo UMELISA SARS-CoV-2 antígeno en todas las muestras estudiadas, teniendo en cuenta la media del cálculo de las fluorescencias. Se obtuvieron diferencias estadísticamente significativas cuando se compararon las muestras positivas y negativas (p<0.0001). Se observan señalados en la figura, los falsos positivos y negativos detectados por el ensayo.
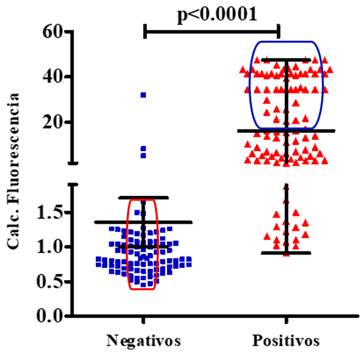
Fig. 1 Análisis del ensayo UMELISA SARS-CoV-2 antígeno en todas las muestras estudiadas, teniendo en cuenta la media del cálculo de las fluorescencias. Se muestra el valor de p de la prueba de Mann-Whitney y señalados los falsos positivos y los falsos negativos.
Se determinó la sensibilidad del UMELISA SARS CoV-2 antígeno en relación a los criterios clínico-epidemiológicos y de laboratorio de las muestras empleadas, los resultados se muestran en la tabla 3.
Tabla 3 Sensibilidad del UMELISA SARS CoV-2 antígeno en relación a los criterios clínico-epidemiológicos y de laboratorio de las muestras empleadas para el análisis
| Criterios clínico-epidemiológicos y de laboratorio de las muestras | Positivos | Sensibilidad % (IC 95 %) |
|---|---|---|
| Muestras positivas a confirmar | 32 | 78,0 (61,9-88,8) |
| Contactos | 36 | 87,8 (72,9-95,4) |
| Evolutivos* | 4 | 100,0 (39,5-97,6) |
| Sospechosos de IRA | 9 | 100,0 (62,8-98,9) |
| Vigilancia | 17 | 65,3 (44,3-82,06) |
| Total | 98 | 80,9 (72,6-87,3) |
* Sin especificar días de evolución; IC 95 %: Intervalos de Confianza del 95 %
Se observa una elevada positividad del ensayo UMELISA SARS CoV-2 antígeno en muestras que provienen del estudio de contactos y particularmente en sospechosos de una infección respiratoria aguda. En el caso de los evolutivos incluidos en este análisis no se conoce de qué día de evolución de la enfermedad fue la colecta de las muestras, en estos casos también la sensibilidad fue del 100 %.
Para conocer la utilidad del ensayo en muestras de casos evolutivos, se analizó la positividad del ensayo a los 5 y 7 días posteriores al primer PCR positivo, cuyos resultados se muestran en la tabla 4. Si tenemos en cuenta el Ct de estos casos (≤33), vemos que la positividad en el caso del 5to. día de evolución se incrementa de 70,6 a 73,3 %. A los 7 días de evolución, la positividad se incrementa de 10 a 12,5 %.
Tabla 4 Positividad del UMELISA SARS CoV-2 antígeno en muestras de casos evolutivos a los 5 y 7 días posteriores al primera PCR positivo
| Días de evolución | Muestras (n) | Positivos (%) |
|---|---|---|
| 5to. día | Total de muestras (17) | 12 (70,6) |
| Muestras con Ct≤33 (15) | 11 (73,3) | |
| 7mo. día | Total de muestras (10) | 1 (10) |
| Muestras con Ct≤33 (8) | 1 (12,5) |
Ct: umbral de ciclo
Discusión
La tecnología SUMA fue desarrollada e implementada en toda la red de salud del país hace más de 3 décadas y es utilizada para el diagnóstico y seguimiento de enfermedades transmisibles y no transmisibles. En el contexto de la epidemia de COVID-19 se incorporan a los ensayos de detección de anticuerpos (UMELISA SARS-CoV-2 IgM, UMELISA ANTI-SARS-CoV-2 y UMELISA SARS-CoV-2 IgG), el UMELISA SARS CoV-2 antígeno, que detecta antígenos correspondientes a la proteína de la nucleocápside viral en muestras de exudado nasofaríngeo procedentes de individuos con sospecha de estar infectados por el virus SARS-CoV-2.
Al comparar los resultados obtenidos en este trabajo con los descritos por el fabricante, observamos que nuestro valor de especificidad (97,2 %) es muy cercano a la especificidad clínica que caracteriza al ensayo según sus productores (97,0 %, IC 95 % 95,5 - 98,0 %), igualmente la sensibilidad obtenida en el presente estudio es similar al planteado por el fabricante, con un valor de 80,9 % (IC 95 % 72,6 - 87,3). Así mismo, la sensibilidad teniendo en cuenta solo las muestras con un Ct ≤ 33, es semejante al expuesto por los productores, donde se plantea un valor incrementado de este parámetro del 81,8 % a 86,3 % para las mismas.12
Los requisitos mínimos de desempeño establecidos por la OMS para los ensayos de detección de antígenos SARS-CoV-2 son poseer una sensibilidad de ≥80 % y una especificidad de ≥97 % comparada con la PCR-TR. Dichos parámetros se cumplen en la evaluación de este ensayo, además, la sensibilidad del ensayo se incrementa en aquellas muestras con Ct≤33, lo que concuerda con que dichas muestras deben poseer mayor carga viral.13
Otro aspecto a destacar en la presente evaluación es en relación a los criterios clínico-epidemiológicos y de laboratorio de las muestras empleadas para el análisis de la sensibilidad, obteniéndose los mejores resultados en aquellos pacientes sospechosos de una IRA, es decir, que poseen síntomas, y también en aquellos que son contactos de casos confirmados. Esta es una de las recomendaciones de uso de las pruebas rápidas de antígenos, priorizando su uso en individuos sintomáticos que cumplen con la definición de caso para COVID-19, y también para individuos asintomáticos con alto riesgo de infección, donde se incluyen a los contactos.13,14
Es importante conocer el comportamiento de los ensayos de detección de antígenos más allá del 5to. día de comienzo de los síntomas, en el caso de UMELISA SARS-CoV-2 antígeno, si bien la positividad no llega a ser ≥80 % de los casos, si fue capaz de detectar el 70,6 % de los casos positivos que incluso se incrementa en muestras con mayor carga viral o con Ct≤33.
Teniendo en cuenta otras investigaciones de ensayos basadas en el mismo principio que UMELISA SARS-CoV-2 antígeno, Osterman et al., estudiaron la validez del ensayo inmunoenzimático semicuantitativo SARS-CoV-2 de Euroimmun Medizinische Labordiagnostika AG (Lübeck, Alemania), que detecta in vitro la proteína N del SARS-CoV-2 a partir de muestras de exudado nasofaríngeo, obteniendo una sensibilidad y especificidad total de 17,76 y 99,67 % respectivamente.7
Así mismo, un estudio llevado a cabo por Domenico et al., en Palermo, para la evaluación de un sistema de prueba de Ag cito-salival SARS-CoV-2 (prueba de laboratorio de Ag COVID-19 de Stark), mostró elevada sensibilidad y especificidad. Además, este método demostró varias ventajas, como la rapidez (30 minutos), el resultado de la reacción antígeno-anticuerpo puede ser directamente visible a simple vista sin necesidad de ser realizado en un laboratorio, sin instrumentación analítica y la de detectar simultáneamente dos antígenos virales específicos, que ciertamente aumenta la especificidad.15
Otro estudio recogido en la literatura se refiere a un ELISA de detección de la nucleoproteína del SARS-CoV-2, pero utilizando muestras de suero o plasma poco tiempo después de la infección (entre 0 y 6 días). En la misma los autores obtuvieron una sensibilidad del 92,9 % y 81,4 % en pacientes hospitalizados y ambulatorios respectivamente. La especificidad fue del 99,8 %. Sin embargo, de 7 a 13 días posteriores la sensibilidad decae a 88,6 % en pacientes hospitalizados. La utilidad de este ensayo estaría en entornos hospitalarios y bancos de sangre, donde se colectan muestras de sangre proporcionando una herramienta de detección simple y económica para el SARS-CoV-2.16 Anteriormente, Che y colaboradores en el 2004, desarrollaron un ELISA de captura para la detección del antígeno N del SARS empleando anticuerpos monoclonales de alta afinidad y utilizando como muestra el suero. Igualmente obtuvieron elevada sensibilidad y especificidad con un pico entre los 6-10 días de la enfermedad.17
Una vez concluida la evaluación del ensayo, en el mes de mayo de 2021 algunos laboratorios de La Habana comenzaron a procesar muestras con el juego de reactivos UMELISA SARS-CoV-2 antígeno, utilizando como muestra el exudado nasofaríngeo. La prueba piloto arrojo una Concordancia entre ambas pruebas de un 97,4 %. Si bien se trata de un estudio controlado, estos resultados nuevamente avalan el buen desempeño clínico de este ensayo.
Recientemente se publicó una guía actualizada de pautas del diagnóstico de COVID-19 en relación a los ensayos de detección de antígenos.18 Si bien los ensayos moleculares continúan siendo el método elegido para diagnosticar la infección por SARS-CoV-2, cuando estas no están disponibles, las pruebas de detección de antígeno ayudan a identificar a las personas con infección por SARS-CoV-2. Poseen elevada especificidad y de baja a moderada sensibilidad, esta última depende de la presencia o ausencia de síntomas, y en pacientes sintomáticos, en el momento de realización de la prueba después de la aparición de síntomas, además, se ha sugerido que repetir la prueba de detección de antígenos aumenta la sensibilidad en comparación con la realización una única vez.
Conclusiones
El ensayo UMELISA SARS-CoV-2 antígeno posee buenas características clínicas acorde con los requisitos establecidos por la OMS para este tipo de prueba. Al ser una tecnología completamente nacional posibilita su extensión y aplicación en la red de laboratorios que realizan diagnóstico y vigilancia epidemiológica de la COVID-19. El ensayo podría ser útil en el seguimiento de pacientes al 5to. día de evolución de la enfermedad.

