










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter v.15 n.2 Ciudad de la Habana Mayo-ago. 1999
Artículos originales
Instituto de Hematología e InmunologíaAportes al estudio de la drepanocitosis. Análisis clínico y hematológico en los primeros 5 años de la vida
Dra. Tania García Peralta,1 Dra. Ileana Nordet Carrera,1 Dr. Sergio Machín García,1 Dr. Alejandro González Otero,1 Lic. Adriana Muñiz Fernández,1 Lic. Gisela Martínez Antuña,1 Dra. Maura Wade Mateo2 y Dra. Eva Svarch1Resumen
Se estudiaron 104 pacientes menores de 6 años de edad. En el 93,1 % el diagnóstico se hizo antes del primer año de la vida y en el 47,6 % fue prenatal. La mediana de seguimiento fue de 31 meses (1-108). La mayor incidencia de eventos clínicos en la anemia drepanocítica (AD) ocurrió entre 1y 3 años. El 91,6 % de las infecciones fueron pulmonares y el 21,7 % de los cuadros respiratorios se catalogaron como síndrome torácico agudo. Los eventos en la hemoglobinopatía SC (HSC) fueron menos frecuentes que en la AD. Sólo 1 paciente falleció en los casi 10 años transcurridos desde que se inició el estudio. La hemoglobina y la hemoglobina fetal (HbF) estuvieron más altas en el sexo femenino en la AD. Los haplotipos del bloque de genes b más frecuentes fueron el Benin y el Bantú. Se constató una frecuencia de a talasemia de 23,8 % en la AD y de 23,07 % en la HSC. No existió correlación entre los haplotipos Bantú y no Bantú y la a talasemia con las manifestaciones clínicas ni con los parámetros hematológicos. El seguimiento sistemático de los pacientes desde temprano en la vida disminuye la mortalidad en este período.Descriptores DeCS: ANEMIA DE CELULAS FALCIFORMES/sangre; ENFERMEDAD DE LA HEMOGLOBINA SC/sangre; BETATALASEMIA/sangre.
La drepanocitosis, que incluye la anemia drepanocítica (AD), la hemoglo-binopatía SC (HSC) y la S b talasemia (Sbtal) y otras combinaciones de la hemoglobina S (HbS) con hemoglobinas anormales, es la enfermedad de carácter genético más frecuente en el mundo.
Su prevalencia es alta en África Ecuatorial, pero se encuentra también en la cuenca del Mediterráneo, en regiones de la India y Arabia Saudita y en las poblaciones de origen africano de los Estados Unidos de Norteamérica, América Central, el Caribe y algunos países de América del Sur. En Cuba, la incidencia del estado del portador (AS) es del 3 % en la población general y del 6 % en la negra y mestiza.1
La drepanocitosis es considerada un problema de salud pública en muchos países.
Aunque su fisiopatología se ha estudiado profundamente, no se conoce en la actualidad ningún tratamiento eficaz e inocuo que prevenga todas las manifestaciones clínicas. Sólo con el trasplante de médula ósea se puede lograr la curación, pero este procedimiento tiene todavía una alta morbiletalidad y costo, por lo que es imposible su aplicación en todos los pacientes.
El objetivo de este trabajo es exponer los datos clínicos y hematológicos de los niños con drepanocitosis atendidos en sus primeros años de vida en la consulta de hemoglobinopatías del Instituto de Hematología e Inmunología (IHI) y correlacionarlos con la hemoglobina fetal (HbF), los haplotipos del bloque de genes b y la a talasemia.
Métodos
En el período comprendido entre junio de 1986 y abril de 1995 se atendieron en la consulta de hemoglobinopatías del IHI, 104 niños menores de 6 años de edad; 53 del sexo masculino y 51 del femenino; 69 con AD, 28 con HSC, 6 con Sb 0tal y con Sb +tal.En la mayoría de los pacientes se realizó el diagnóstico prenatal y fueron enviados por los médicos de la consulta de Genética del Hospital Ginecoobstétrico «Ramón González Coro».
No se estudió ningún niño con más de 2 años de edad en el momento de la primera consulta. El seguimiento fue mensual en el primer año de edad y después cada 3 meses.
En cada consulta se realizó un interrogatorio minucioso sobre las manifestaciones clínicas ocurridas en el intervalo entre las mismas.
En los enfermos en quienes no se hizo el diagnóstico prenatal y por lo tanto, comenzaron a ser atendidos en nuestra consulta en diferentes etapas de su vida, en el primer encuentro se llevó a cabo un interrogatorio exhaustivo sobre todos los eventos que ocurrieron en el período anterior. No se incluyó a ningún paciente en el que no se hubiera podido determinar el cuadro clínico anterior a su primera consulta.
En cada entrevista se realizó un examen físico cuidadoso. A partir de los 3 meses de edad se administró ácido fólico (5 mg al día) y penicilina oral profiláctica (125 mg 2 veces al día) a los niños menores de 3 años; en los niños entre 3 y 5 años, la dosis de la penicilina oral fue de 250 mg 2 veces al día.
En cada consulta se realizó una labor de educación y en la primera entrevista se le entregó a la madre un folleto realizado en nuestra institución en el que se explican las características más importantes de la enfermedad y su manejo. Se enseñó a la madre a palpar el bazo y a que acudiera al centro asistencial si notaba un aumento de tamaño de éste, acentuación de la palidez, irritabilidad y fiebre.
Se insistió en la importancia de la ingestión abundante de líquidos y de evitar esfuerzos físicos excesivos, así como los cambios bruscos de temperatura.
Los exámenes de laboratorio de realizaron por las técnicas habituales y la HbF por el método de Betke.2
En 96 pacientes se determinaron los haplotipos del bloque de genes b3 (se excluyeron de este estudio los haplotipos sanos) y en 35 los genes a.4
El hemograma y los reticulocitos se hicieron en cada consulta y la HbF se realizó anualmente. Todas las investigaciones de laboratorio se llevaron a cabo en condiciones basales, sin crisis en los 15 días previos ni transfusiones en los 3 meses anteriores.
Para procesar los datos se utilizó el paquete estadístico para computadora Epinfo versión 5 y el Statgraf versión 2.3. Se emplearon los siguientes intervalos de edad en niños: < 1, 1, 2, 3, 4 y 5.
El análisis estadístico se realizó por medio del cálculo de las distribuciones de frecuencia de las variables cualitativas: sexo, haplotipos y genes a. Se correlacionaron los haplotipos divididos en 2 grupos: Bantú y no Bantú; y los genes a con los eventos clínicos, las cifras de hemoglobina y HbF, por medio del coeficiente de correlación de Pearson.
Para las variables cuantitativas se calculó la media (X) y la desviación estándar (DE). Se realizó un resumen donde se contabilizó el total de eventos clínicos acumulados por cada paciente desde el nacimiento hasta la última consulta.
Se aplicó el análisis de varianza (Anova), clasificación simple o la prueba de Kruskell-Wallis5 para comparar los grupos con AD y HSC. No se incluyó la Sbtal por el escaso número de pacientes. El nivel de significación fue del 5 % en todos los casos.
Resultados
En el 93,1 % de los pacientes se diagnosticó la enfermedad en el primer año de la vida, en el 47,6 % éste fue prenatal y el 55,8 % acudió a la primera consulta en ese período.La mediana de seguimiento fue de 31 meses (1-108).
En la tabla 1 se muestra la media y la desviación estándar de los eventos clínicos por año de edad en la AD. En los niños menores de 1 año hubo una cantidad menor de eventos que en el resto. En todos se encontraron diferencias significativas, con una incidencia mayor de 1 a 3 años en la mayoría de ellos. El 91,6 % de las infecciones fueron pulmonares y el 5,6 % fueron episodios febriles de más de 7 días de duración, sin signos de localización. El 21,7 % de los cuadros respiratorios agudos se catalogaron como síndrome torácico agudo. Las crisis mano-pie disminuyeron a los 4 años y no se produjeron a los 5. Tampoco se comprobaron crisis de secuestro esplénico a los 4 y 5 años y probablemente por esta razón se registraron pocas transfusiones es ese lapso.
| | |||||||||||||||||||
| | | | | | | ||||||||||||||
| Eventos | DE | DE | DE | DE | DE | DE | p | ||||||||||||
| Ingresos |
| | | | | | | | | | | | | ||||||
| Infecciones | | | | | | | | | | | | | | ||||||
| Crisis vasooclu- | |||||||||||||||||||
| sivas | | | | | | | | | | | | | | ||||||
| Crisis mano-pie | | | | | | | | | | | | | | ||||||
| Crisis de secues- | |||||||||||||||||||
| tro esplénico | | | | | | | | | | | | | | ||||||
| Transfusiones | | | | | | | | | | | | | | ||||||
Los ingresos, las manifestaciones clínicas y las transfusiones, fueron menos frecuentes en la HSC que en la AD. El 98,7 % de los procesos infecciosos fueron pulmonares y el diagnóstico de síndrome torácico agudo se planteó en el 10 % de los niños. Se produjeron pocas crisis mano-pie y sólo un paciente tuvo una crisis de secuestro esplénico. Hubo variaciones entre los años, que no mostraron diferencias significativas, salvo las transfusiones.
En la Sb tal no hubo pacientes menores de un año ni de 4 y 5 años; los niños de un año no tuvieron manifestaciones clínicas. No se produjeron crisis de secuestro esplénico ni se indicaron transfusiones a los 2 años de edad. A los 3 años no se registraron crisis vasooclusivas (CVO) ni crisis mano-pie.
Un resumen de todos los eventos en los primeros 5 años de la vida se presenta en la figura 1. En ninguno de los 104 pacientes se presentó CVO del SNC en el período del estudio.

Fig. 1: Porcentaje de pacientes que presentaron eventos clínicos en la drepanocitosis.
Los valores de hemoglobina en el sexo femenino en los pacientes con AD estuvieron significativamente más elevados que en el masculino (tabla 2). También se encontraron diferencias significativas entre los años en el sexo femenino. No hubo diferencias entre los sexos en lo que respecta a los reticulocitos (tabla 2). A pesar de que la HbF se encontró más elevada en el sexo femenino, las diferencias por año de edad no fueron significativas (fig. 2). Sin embargo, cuando se compararon los valores globales de HbF: 11,8 ± 12,4 % en el sexo masculino y de 18,6 ± 12,7 % en el femenino, la diferencia fue estadísticamente significativa (p = 0,008).
| Hemoglobina | Reticulocitos | HbF | ||||||||||||||
| (g/dL) | (%) | (%) | ||||||||||||||
| Edad | Masculino | Femenino | Masculino | Femenino | Masculino | Femenino | ||||||||||
| (años) | |
| |
| | |
| | | | | | | | | |
| < 1 | | | | | | | | | | | | | | | | |
| 1 | | | | | | | | | | | | | | | | |
| 2 | | | | | | | | | | | | | | | | |
| 3 | | | | | | | | | | | | | | | | |
| 4 | | | | | | | | | | | | | | | | |
| 5 | | | | | | | | | | | | | | | | |
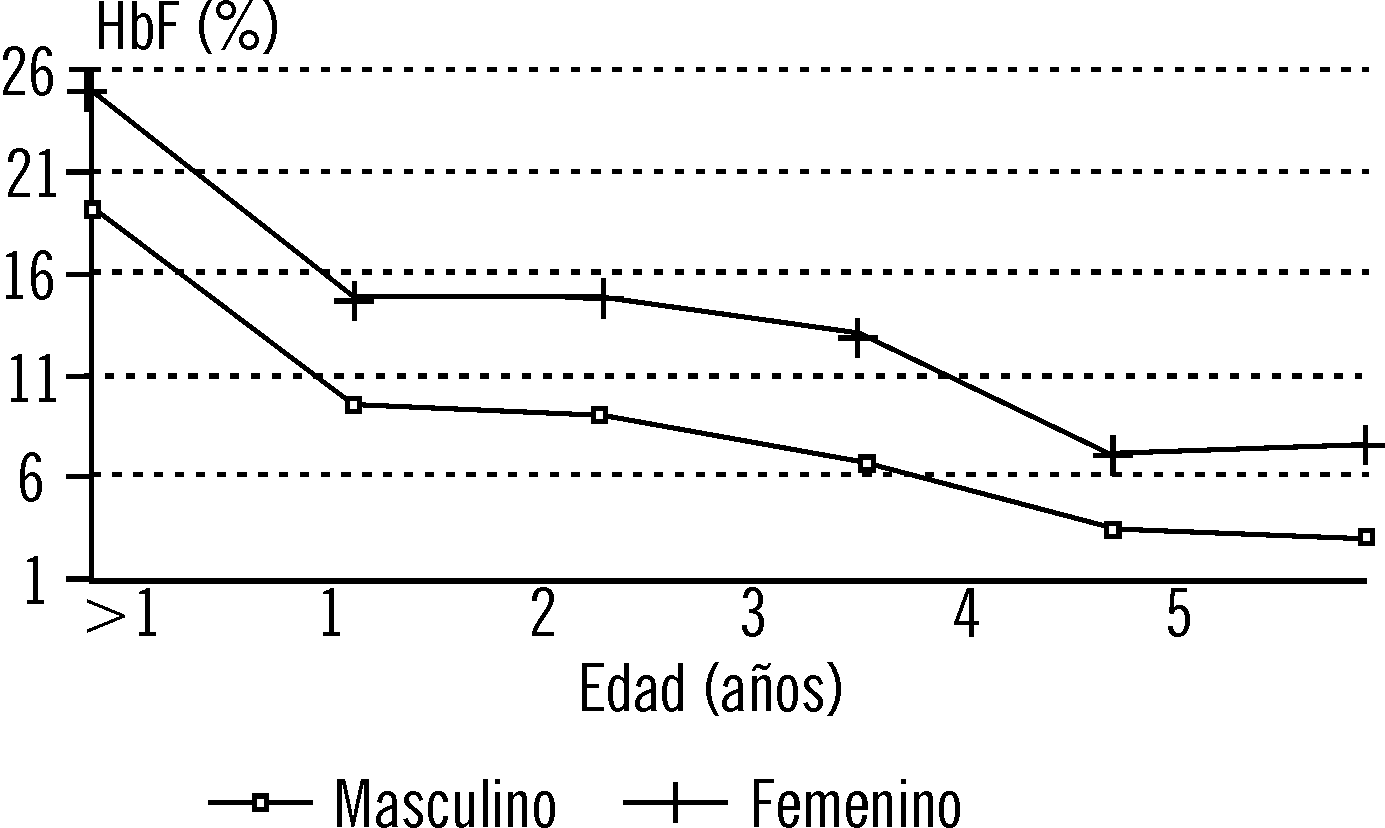
Fig. 2. Hemoglobina fetal (%) por edad y sexo en la anemia drepanocítica.
No se encontraron diferencias entre los sexos ni entre los años, en cuanto a las cifras de leucocitos y plaquetas (tabla 3).
| Leucocitos | Plaquetas | ||||
| (x 109/L) | (x 109/L) | ||||
| Edad (años) | DE | ||||
| < 1 | 10,2 | 2,7 | 348 | ||
| 1 | 11,8 | 3,8 | 309 | ||
| 2 | 13,4 | 5,4 | 328 | ||
| 3 | 14,0 | 3,0 | 348 | ||
| 4 | 13,3 | 3,7 | 336 | ||
| 5 | 11,9 | 2,4 | 3 | ||
| p | 0,07 | 0,1 | |||
En la tabla 4 se observan los datos hematológicos en la HSC. No existieron diferencias significativas entre los sexos ni edades, salvo en la HbF, que como era de esperar, disminuyó con la edad. Contrariamente a lo que ocurrió en la AD, el porcentaje global de la HbF no mostró diferencias entre los sexos.
Tabla 4. Hemoglobina, reticulocitos, HbF, leucocitos y plaquetas en 28 niños con hemoglobinopatía SC
| | |||||||||||||
| | | | | | |||||||||
| Parámetros | |
| | | |
| |
| |
| |
| |
| Hemoglobina (g/dL) | | | | | | | | | | | | | |
| Reticulocitos (%) | | | | | | | | | | | | | |
| HbF (%) | | | | | | | | | | | | | |
| Leucocitos (x 109/L) | | | | | | | | | | | | | |
| Plaquetas (x 109/L) | | | | | | | | | | | | | |
En la Sbtal hubo muy pocas observaciones.
Los haplotipos del bloque de genes b se muestran en la figura 3. Los más frecuentes fueron el Benin y el Bantú.
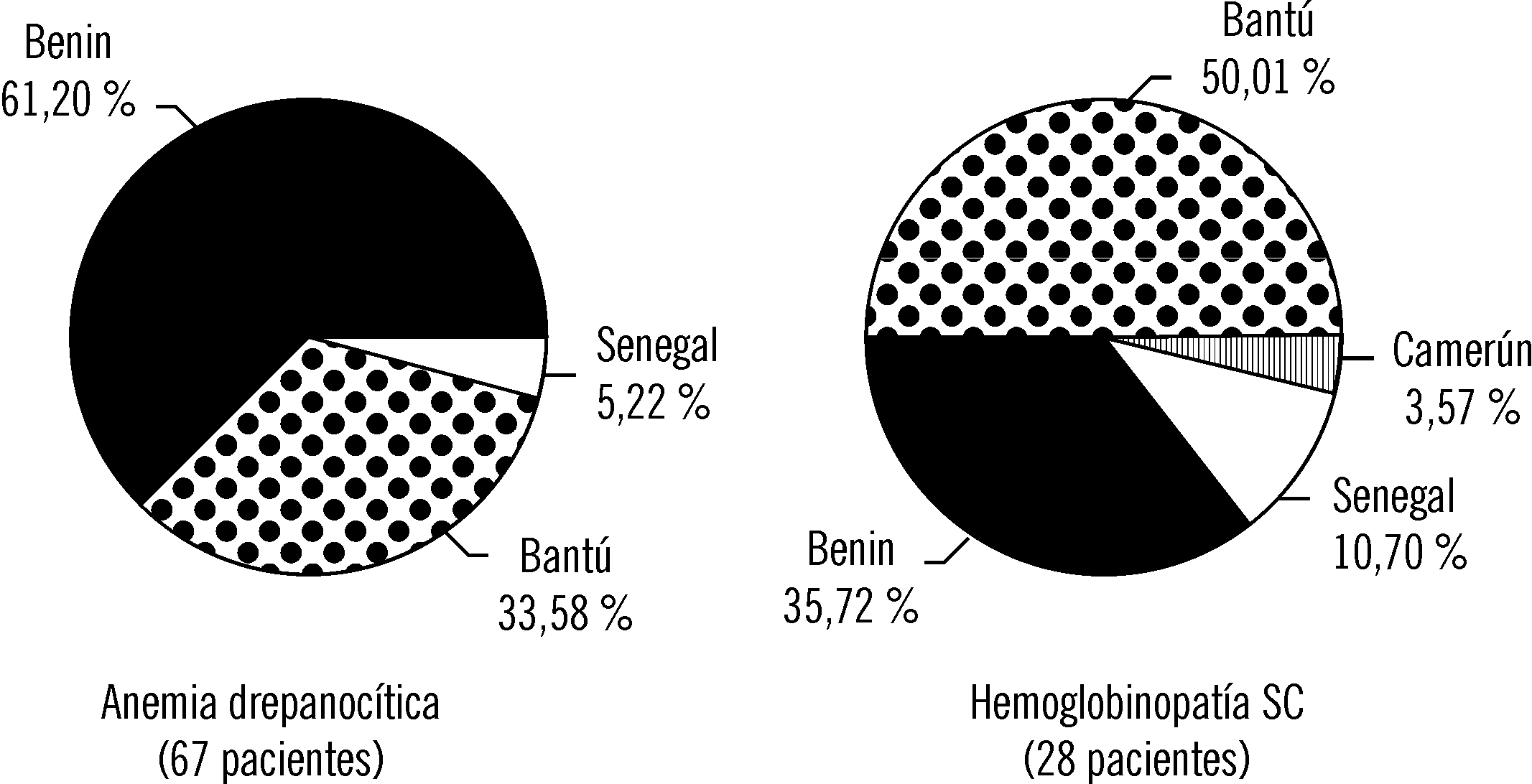
Fig. 3. Haplotipos del bloque de genes b n la drepanocito-sis.
La frecuencia de a talasemia 2 en la AD fue del 23,8 % y en la HSC del 23,07 %. Sólo 1 niño con AD y 1 con HSC tuvieron a talasemia 1. En ninguno con Sb tal se comprobó la coexistencia de a talasemia. Los haplotipos Bantú y no Bantú y los genes a no se correlacionaron de manera significativa con ningún evento clínico ni con los parámetros hematológicos.
Discusión
En el cuadro clínico del niño pequeño predominan los episodios agudos: infecciones, crisis mano-pie, crisis de secuestro esplénico y son poco frecuentes las lesiones de los órganos ocasionadas por la falciformación crónica.En esta investigación, las manifestaciones clínicas de la AD fueron menos frecuentes en el niño menor de 1 año de edad, lo que se debe al efecto protector de la HbF en los primeros meses de la vida.1 En los años siguientes se encontraron diferencias significativas en todos los eventos en cada año y se observó un aumento de éstos entre 1 y 3 años. No es posible comparar estos datos con otras observaciones, ya que hasta donde conocemos, no existen en la literatura trabajos en los que se haya analizado el cuadro clínico por año de edad; sin embargo, se ha descrito una mortalidad más elevada entre 1 y 3 años.6
El número de ingresos refleja la severidad del cuadro clínico. Un estudio realizado en Inglaterra en 113 pacientes con AD entre 1 y 17 años mostró un promedio anual de ingresos de 0,53.7 Los promedios en este trabajo fueron algo más bajos, lo que puede deberse al rango diferente de edades.
Las infecciones constituyen la manifestación clínica de mayor incidencia en el niño pequeño6 y la causa más frecuente de muerte en este período de la vida.1 El germen más involucrado es el neumococo y la profilaxis con penicilina ha disminuido el número y gravedad de las infecciones producidas por este microorganismo.8 No encontramos datos en la literatura sobre el promedio anual de infecciones en los primeros años de la vida, pero el porcentaje de nuestros enfermos que presentaron esta complicación es semejante al descrito por otros autores6 y ningún paciente falleció por esta causa.
Posiblemente a consecuencia de la profilaxis con penicilina han aparecido cepas de neumococo resistentes a este antibiótico, e incluso, resistentes a múltiples antimicrobianos, por lo que se aconseja utilizar la vacuna antineumocóccica con reinmunización a los 6 u 8 años y cambiar la política de antibióticos utilizando una cefalosporina de tercera generación y vancomicina cuando se diagnostica una infección grave en áreas en las que se conoce que la resistencia del neumococo a la penicilina es prevalente.9
Se ha planteado que la causa más frecuente de síndrome torácico agudo en el niño pequeño es la infección, ya que estos pacientes no han desarrollado aún anticuerpos contra muchos microorganismos.10 Sin embargo, cuidadosos estudios bacteriológicos y virales excluyeron la etiología infecciosa en la mayor parte de los niños.11
El número de CVO en el niño es muy variable.12 En el presente trabajo, el porcentaje de niños que las presentaron es semejante al descrito por otros autores.6 Recientemente se ha señalado que las CVO influyen en el daño de los órganos, e incluso, en la sobrevida.13
En nuestros pacientes, la crisis mano-pie fue frecuente al año y 2 años de edad, y el porcentaje de niños que la padecieron es semejante al que comunican en Jamaica (alrededor del 40 %).14
La crisis de secuestro esplénico constituye la segunda causa de muerte en los niños menores de 5 años.15 En este estudio, el número de enfermos con esta complicación fue mayor que en otros,16 pero esto se debe a que muchos niños pequeños fueron remitidos a nuestro servicio por crisis de secuestro esplénico para valorar esplenectomía parcial y posteriormente, se incluyeron en el seguimiento.
El porcentaje de enfermos que recibieron transfusiones de glóbulos rojos es semejante al que describen otros autores.17 Sus indicaciones en nuestros pacientes son muy precisas: crisis aplástica o de secuestro, infecciones graves y previas a la cirugía con anestesia general, fundamentalmente.
La mortalidad en la AD en los primeros años de la vida hace pocos años era del 15 %.6 En la actualidad es del 2,6 % y la sobrevida global alcanza la cuarta década de la vida.13 Sólo falleció 1 niño en los más de 9 años transcurridos desde que se inició esta investigación.
En la HSC se encontraron menos eventos clínicos que en la AD, tal como lo señalan en la literatura16 y éstos aparecen más tardíamente. El 22 % se mantiene asintomático en la primera década de la vida18 y la sobrevida global alcanza la cuarta y la quinta décadas.6
En nuestro estudio el número de infecciones en la HSC fue algo menor que en la AD. Existen controversias en cuanto a si las infecciones están aumentadas en la HSC. Algunos autores no lo demuestran y no aconsejan la profilaxis con penicilina,19 mientras que otros no encuentran diferencias entre la HSC y la AD.20
Los valores de hemoglobina de nuestros pacientes fueron semejantes a los que se encontraron en otras series.21 Se mantuvieron estables en los diferentes años en el sexo masculino y hubo algunas variaciones entre los años en el sexo femenino, en el cual las cifras se encontraron más elevadas que en el masculino en todas las edades. Estos hallazgos concuerdan con lo que se describe en otros trabajos21 y posiblemente esté en relación con una HbF más elevada en las niñas.22
Las cifras de reticulocitos en la AD estuvieron dentro del rango que se describe en la literatura (del 10 al 20 %).23 No existieron diferencias entre los años ni entre los sexos.
Cuando analizamos los valores de HbF por año de edad no se demostraron diferencias entre los sexos, a pesar de que fueron más elevados en el sexo femenino. La HbF depende de la edad, de los haplotipos del bloque de genes b y del número de genes a, pero los factores más importantes parecen ser: un locus ligado al cromosoma X que regula la producción de glóbulos rojos con HbF (células F) y los haplotipos.22
En este trabajo se comprobó un número elevado de leucocitos en todas las edades, lo que concuerda con otros estudios, en los que se plantea que la leucocitosis puede deberse a estimulación de la célula progenitora, asplenia o demarginación.24
Las plaquetas estuvieron dentro del rango normal en nuestro laboratorio. No hubo diferencias significativas en los leucocitos y plaquetas entre las edades ni entre los sexos.
En la HSC las cifras de hemoglobina y reticulocitos fueron semejantes a las de otros trabajos.25 La HbF se encontró más baja que en la AD y no se detectaron diferencias entre los sexos. Los leucocitos y las plaquetas estuvieron dentro del rango normal, lo que puede deberse a la menor intensidad del proceso hemolítico en esta hemoglobinopatía.25
En un estudio reciente realizado en Cuba en adultos, en la AD se demostró una disminución del haplotipo Bantú con la edad y un aumento del Senegal, lo que no ocurrió en la HSC. Esto puede reflejar la sobrevida menor de los pacientes con el haplotipo Bantú, en dependencia de la edad y del tipo de drepanocitosis.26 La ausencia de correlación en nuestros enfermos, entre las manifestaciones clínicas y hematológicas y los haplotipos, puede deberse a que el haplotipo Bantú influye sobre todo en el daño irreversible de los órganos que se produce en el adulto.27
No se encontró en la presente investigación una correlación entre la a talasemia y el cuadro clínico y hematológico, aunque el número de niños estudiados fue pequeño y algunos autores señalan que la a talasemia mejora las manifestaciones clínicas y hematológicas en la AD.28
A pesar de los conocimientos profundos sobre los aspectos genéticos, la fisiopatología y el cuadro clínico de la drepanocitosis, no existe todavía un tratamiento eficaz e inocuo para prevenir o tratar las múltiples complicaciones de la enfermedad.
Algunas propuestas terapéuticas reentes son atractivas. La hidroxiurea disminuye el número de CVO tanto en adultos29 como en los niños.30 En el futuro existe la posibilidad de aplicar terapia génica, pero en la actualidad el único tratamiento curativo es el trasplante de médula ósea, que por su complejidad y alto costo sólo es aplicable en pacientes con un cuadro clínico grave y con un hermano HLA compatible.31
La mejoría de la calidad de la vida y la disminución de la mortalidad por la drepanocitosis en Cuba, no se deben solamente a una atención médica adecuada, sino también a una mejoría en las condiciones socioeconómicas y en la nutrición de los pacientes.
Summary
104 patients under 6 were studied. In 93,1 % of them the diagnosis was made before the first year of life, whereas in 47.6 % it was prenatal. The median of follow-up was 31 months (1-108). The highest incidence of clinical events in sickle cell anemia (SCA) occurred between 1 and 3 years old. 91.6 % of the infections were pulmonary and 21.7 % of the respiratory episodes were considered as acute chest syndrome. The events in hemoglobinopathy SC (HSC) were less frequent than in sickle cell anemia. Only one patient died in a period of almost 10 years since the study began. Hemoglobin and fetal hemoglobin (HbF) were higher among females with SCA. The commonest haplotypes of the block of genes b were Benin and Bantu. It was observed a frequency of thalassemia of 23.8 % in sickle cell anemia and of 23.7 % in HSC. Bantu and non-Bantu haplotypes and a thalassemia were not associated with the clinical manifestations or with the hematological parameters. The systematic follow-up of the patients since the first years of life reduces mortality in this period.Subject headings: ANEMIA, SICKLE CELL/blood; HEMOGLOBIN C/blood; BETA-THALASSEMIA/blood.
Referencias Bibliográficas
1. Colombo B, Svarch E, Martínez G. Genética y clínica de las hemoglobinas humanas. La Habana: Editorial Pueblo y Educación, 1994.
2. Betke K, Martín H, Schlicht I. Estimation of small percentages of foetal hemoglobin. Nature 1959;184:1877.
3. Nagel RL, Rao SK, Dunda-Belkhodja O, Connolly MM, Fabry ME, George A, et al. The hematological characteristics of sickle cell anemia bearing the Bantu haplotype: the relationship between G and HbF level. Blood 1987;69:1026-30.
4. Higgs D, Aldridge B, Lamb J, Hayes RJ, Grandison I, Lowrie I, et al. The interaction of alpha thalassemia and homozygous sickle cell disease. N Engl J Med 1982;306:1441-6.
5. Still R, Torrie J. Principles and procedures of statistics. New York:McGraw Hill, 1960:406.
6. Powars DR. Natural history of sickle cell disease. The first ten years. Semin Hematol 1975;12:267-85.
7. Murtaza LN, Strovd CE, Davis LR, Cooper DJ. Admissions to hospital of children with sickle cell anaemia: a study in South London. Br Med J 1981;282:1048-51.
8. Pegelow CH, Armstrong FD, Light S, Toledano SR, Davis J. Experience with the use of prophylactic in children with sickle cell anemia. J Pediatr 1991;118:736-8.
9. Leggiadro RJ. The clinical impact of resistance in the management of pneumococcal disease. Infect Dis Clin North Am 1997;11:867-74.
10. Castro O, Brambilla DJ, Thorington B, Reindorf CA, Sott RB, Gillete Vera JC, et al. The acute chest syndrome in sickle cell disease. Incidence and risk factors. Blood 1994;84:643-9.
11. Sprinkle RH, Cole T, Smith S, Buchanan GR. Acute chest syndrome in children with sickle cell disease. A retrospective analysis of 100 hospitalized cases. Am J Pediatr Hematol 1986;8:105-10.
12. Vishinsky EP. Comprehensive care in sickle cell disease: its impact on morbidity and mortality. Semin Hematol 1991;28:220-6.
13. Platt OS, Brombilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 1994;330:1639-44.
14. Hvo MH, Friedlaender GE, Marsh JS. Orthopoedic manifestations of sickle cell disease. Yale J Biol Med 1990;63:195-207.
15. Emond A, Collis R, Darvill D, Higg DR, Maude Gtl, Serjeant GR. Acute splenic sequestration in homozygous sickle cell disease: natural history and management. J Pediatr 1985;107:201-6.
16. De Montalembert M, Guilloud-Bataille M, Feinglod J, Girot R. Epidemiological and clinical study of sickle cell disease in France, French Guiana and Algeria. Eur J Haematol 1993;51:136-40.
17. Luban N. Variability in rates of alloinmunization in different groups in children with sickle cell disease: effect of ethnic back ground. Am J Pediatr Hematol Oncol 1989;11:314-9.
18. William S, Maude GH, Serjeant GR. Clinical presentation of sickle cell hemoglobin C disease. J Pediatr 1986;109:586-9.
19. Roger ZR, Buchanan GR. Bacteremia in children with sickle cell hemoglobin C disease and sickle b+ thalassemia: is prophylactics penicillin necessary? J Pediatr 1995;127:348-51.
20. Pearson HA, Gallagher D, Chilcote R, Sulliran E, Willimas J, Espeland M, et al. Developmental pattern of splenic dysfunction in sickle cell disorders. Pediatrics 1985;76:392-7.
21. Serjeant GR, Grandison Y, Lowrie Y, Vaidya S, Mason KP, et al. The development of haemotological changes in homozygous sickle cell disease: a cohort study from birth to 6 years. Br J Haematol 1981;48:533-43.
22. Chang YC, Maier-Redelsperger M, Smith KD, Contul DR, De Montalembert M. The relative importance of X-linked FCP locus and b-globin haplotypes in determining haemoglobin F levels: a study of SS patients homozygous for S haplotypes. Br J Haematol 1997;96:806-14.
23. Hayes RJ, Beckford M, Grandison Y, Mason K, Serjeant GR. The haematology of the steady state homozygous sickle cell disease: frequency distributions, variations with age and sex, longitudinal observation. Br J Haematol 1985;59:369-82.
24. West MS, Wethers D, Smith J, Steinberg M. The Cooperative Study of Sickle Cell Disease. Laboratory profile of sickle cell disease: a cross-sectional analysis. J Clin Epidemiol 1992;45:893-909.
25. Serjeant GR. Sickle cell disease. Oxford: University, 1985;289-95.
26. Muñíz A, Corral L, Aláez C, Svarch E, Espinosa E, Carbonell N, et al. Sickle cell anemia and b-gene cluster haplotype in Cuba. Am J Hematol 1995;49:163-4.
27. Nagel R, Fabry M, Pagrier J. Hematological and genetic markers in sickle cell disease. Semin Hematol 1991;28:180-201.
28. Nagel RL. Severity, pathobiology, epistatic effect and genetic markers in sickle cell anemia. Semin Hematol 1991;28:180-201.
29. Charache S, Teerin ML, Moore RD, Dover GJ, Barton FB, Eckerts SV, et al. Study of hydroxyurea in sickle cell anemia. N Eng J Med 1995;332:1317-32.
30. Rogers ZR. Hydroxyurea therapy for diverse pediatric population with sickle cell disease. Semin Hematol 1997;34:42-7.
31. Walters MC, Patience M, Leisenring W, Eckman JR, Scott JP, Mentzer WC, et al. Bone marrow tranplantion for sickle cell disease. N Engl J Med 1996;335:369-76.Recibido: 10 de diciembre de 1998. Aprobado: 14 de diciembre de 1998.
Dra. Tania García Peralta. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800, Ciudad de La Habana, Cuba. Teléfono (537)578268. Fax (537)338979. e-mail:ihidir@hemato.sld.cu

