










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter v.17 n.2 Ciudad de la Habana Mayo-ago. 2001
Artículos de revisión
Instituto de Hematología e Inmunología
Estado actual del mecanismo de la coagulación sanguínea
Lic. Alina Díaz Concepción y Dra. Delfina Almagro Vázquez
Resumen
Actualmente se acepta de forma categórica que el evento iniciador principal de la coagulación sanguínea es la exposición del factor tisular (FT), lo cual da lugar a la formación del complejo factor VIIa/FT que activa a los factores IX y X en la superficie de las células que expresan el FT, y se forman las primeras cantidades de trombina. Esta trombina ejerce múltiples funciones en el mecanismo hemostático, pero es insuficiente para lograr una hemostasia eficaz, lo cual solo se logra con el ensamblaje del complejo protrombinasa en la superficie plaquetaria. El descubrimiento en la década de los 90 de la activación del factor XI por la trombina, permite explicar las observaciones clínicas en los pacientes con deficiencia de los factores de la fase de contacto de la coagulación: estos no presentan complicaciones hemorrágicas (excepto el déficit de factor XI). El descubrimiento realizado recientemente de la activación de la precalicreína y del factor XI por una cisteín proteasa localizada en la superficie de las células endoteliales fundamentalmente hace sospechar un nuevo papel de estas proteínas in vivo. La coagulación sanguínea es un proceso estrechamente regulado, y por su significación fisiológica es importante la regulación de la vía del factor tisular que limita las cantidades iniciales de trombina, así como la regulación de las proteasas formadas durante el mecanismo de la coagulación por la antitrombina III y de los cofactores activados por el sistema de la proteína C.
DeCS: FACTORES DE COAGULACION SANGUINEA/análisis; PROTROMBINA/análisis.
El conocimiento actual acerca del mecanismo de la coagulación sanguínea es el resultado de muchas décadas de observaciones clínicas y de experiencias de laboratorio, que en los últimos años se ha acrecentado aceleradamente gracias a los avances en las tecnologías de purificación de proteínas, el cultivo de tejidos y la aplicación en este campo de los avances de la biología molecular. Clásicamente se ha interpretado el mecanismo de la coagulación mediante la hipótesis de la cascada enzimática, distinguiéndose 2 vías: la vía intrínseca activada cuando la sangre se pone en contacto con determinadas superficies, y la vía extríseca iniciada cuando se produce un daño tisular. Ya desde la década de los 70, se aceptó que el mecanismo de la coagulación es el resultado de la interacción de varios compartimentos macromoleculares que involucran un zimógeno y una serín proteasa vitamina K dependientes, asociados con cofactores ensamblados sobre una membrana1 (fig.1). Son esenciales para la formación de esos complejos los factores vitamina K dependientes, la vitamina K es necesaria para la conversión de las proteínas precursoras de los factores II, VII, IX y X en proteínas con actividad coagulante y de los inhibidores proteína C, y proteína S en proteínas con actividad biológica,2 lo cual ocurre mediante la modificación postraslacional en residuos específicos de ácido glutámico, resultando en la formación de ácido g carboxiglutámico (Gla); la enzima que cataliza esta reacción, la g carboxilasa, usa un derivado de la vitamina K como cofactor. Este cambio estructural relativamente mínimo aumenta las cargas negativas de la molécula y permite la unión con los iones Ca2+.3 El enlace al Ca2+ de los residuos Gla induce los cambios conformacionales requeridos para el enlace de estos factores de la coagulación a las membranas,4 los iones Ca2+ sirven también como puentes entre los residuos Gla cargados negativamente y los grupos fosfato de los fosfolípidos, y por otra parte, se han propuesto interacciones más específicas del módulo Gla con las cabezas fosfolipídicas.5 Los complejos formados en el proceso de la coagulación sanguínea comparten muchas características comunes, primero son funcionalmente análogos con constituyentes estructurales homólogos, la enzima del complejo exhibe requerimientos similares para el ensamblaje y la actividad, y en cada caso, la formación del complejo enzimático conduce a un aumento significativo de 105 a 106 veces en la velocidad de hidrólisis del sustrato.6
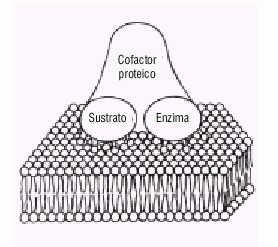
Fig. 1. Composición de los complejos formados durante la coagulación sanguínea.
Sistema de contacto de la coagulación
Por más de 25 años, la mayoría de los investigadores han aceptado la hipótesis que plantea que el factor XII inicia la activación del sistema de contacto de la coagulación por enlace con superficies negativamente cargadas, lo cual es la base para los ensayos de la coagulación que se fundamentan en la activación por contacto. También este parece ser el mecanismo por el cual este sistema es activado in vivo cuando la sangre se expone a superficies artificiales, tal como ocurre en intervenciones quirúrgicas o en pacientes con válvulas cardíacas artificiales.7
En la activación por contacto participan los factores XII, XI, la precalicreína y el quininógeno de alto peso molecular (QAPM). La etapa inicial de la vía intrínseca no es comprendida completamente; se plantea que la unión del factor XII a una superficie cargada negativamente, induce un cambio conformacional en la molécula que desencadena un mecanismo de autoactivación, o que facilita su activación por otras enzimas activadas que se encuentran circulando de forma permanente.8,9
El factor XIIa actúa proteolíticamente sobre el factor XI y la precalicreína, estas circulan en fase fluída unidas al QAPM, lo que les permite unirse con las superficies cargadas negativamente para su posterior reacción con el factor XIIa.10 La calicreína formada activa al factor XII en una reacción que es 100 veces más rápida que la auto-activación.
El factor XI a activa al factor IX,11 el factor IXa resultante de esta reacción es una serín proteasa vitamina K dependiente que cataliza la activación del factor X en presencia de fosfolípidos, iones calcio y factor VIIIa como cofactor no enzimático. Su función es aumentar la kcat de la reacción en varios órdenes de magnitud, y la superficie fosfolipídica parece contribuir a este efecto sobre la kcat.12
Es bien conocido, sin embargo, que las deficiencias de factor XII, precalicreína y QAPM no están asociadas con el sangramiento, aun cuando ellas prolongan marcadamente los tiempos de coagulación de las pruebas dependientes de superficies de contacto, lo que sugiere que este sistema contribuye poco a la hemostasia.13,14 Por otra parte, no se ha descrito convincentemente una superficie que in vivo pueda iniciar la activación de este sistema. Se ha sugerido que el ensamblaje y la activación del sistema de contacto puede llevarse a cabo en membranas biológicas de células endoteliales, plaquetas, neutrófilos y monocitos,15,16 además la autoactivación del factor XII pude tomar horas en dependencia de la superficie. Estos argumentos son contrarios a asignarle un papel preponderante al sistema de contacto, al menos como iniciador del mecanismo de la coagulación in vivo. Recientemente, en un intento de esclarecer el papel de estas proteínas in vivo, se ha formulado una hipótesis para explicar el ensamblaje y la activación de este sistema en membranas celulares. Dicha hipótesis es una alternativa al mecanismo propuesto de autoactivación del factor XII. Diferentes resultados experimentales indican la existencia de un receptor proteico o receptores para el QAPM en células endoteliales.17-21 Usando una columna de afinidad preparada con QAPM como ligando, Hasan y otros22 purificaron una proteína de 54 kd en lisados de células endoteliales que fue identificada como citoqueratina-1; el antígeno de esta proteína se ha encontrado además en plaquetas y granulocitos, lo que indica que la citoqueratina-1 puede servir como sitio de enlace para el QAPM en esas células. El QAPM se enlaza a la citoqueratina 1 natural o recombinante solo en presencia de iones Zn2+.
Otras proteínas pueden participar como sitios de enlace o receptores alternativos para el QAPM. Se ha demostrado que este se enlaza al receptor del activador del plasminógeno tipo uroquinasa (R-Apu) en la misma región de enlace de la vitronectina.21 El R-Apu está presente en células endoteliales y granulocitos, pero su epítope no se ha encontrado en plaquetas, por lo tanto, él solo no puede explicar todos los enlaces del QAPM con las células. Existen evidencias indirectas de que el QAPM interactúa al menos con 2 proteínas más en las plaquetas: la glicoproteína IX-V y un receptor activable por proteasas.22,23 Se ha demostrado que la calicreína enlazada al QAPM unido con las células endoteliales probablemente es activada por una cisteínproteasa no típica que requiere de iones divalentes para su actividad,7 particularmente de iones Zn2+.
En células endoteliales la activación de la precalicreína inicia el sistema, contrario a lo que ocurre en superficies artificiales, donde inicia el sistema la activación del factor XII, lo que sugiere una nueva función de la precalicreína en las membranas biológicas.24 La activación del factor XI en células endoteliales puede ocurrir en el plasma normal o en plasma deficiente de factor XII, pero no en plasma deficiente de precalicreína, y parece ocurrir por un mecanismo similar a la activación de la precalicreína, que requiere la presencia de QAPM y una concentración óptima de iones Zn2+ de 7 a 10 µM, que es independiente de la autoactivación, la a-trombina o el factor XIIa.20
Vía alternativa de la coagulación
La vía extrínseca, llamada actualmente vía alternativa, se inicia con el contacto de la sangre con el factor tisular (FT). El FT es una proteína presente en células endoteliales, monocitos y macrófagos, en el tejido extravascular especialmente en la adventicia, en el epitelio y mucosas, en astrocitos en el cerebro y en el estroma de células del endometrio.25,26 Está constituida por 263 aminoácidos organizados en 3 dominios: un dominio extravascular de 219 aminoácidos, un dominio transmembrana de 23 aminoácidos y un dominio intracelular de 21 aminoácidos.27 El FT se encuentra disponible en la cara apical de las células endoteliales vasculares. La expresión polarizada del FT permite que el factor VIIa interactúe específicamente con aquellas células endoteliales que han sido suficientemente dañadas o estimuladas para expresar el FT. La síntesis del FT puede ser inducida por endotoxinas, inmunocomplejos, ésteres de formol, trombina, interleucina-1 y factor de necrosis tumoral, entre otros.25 Aproximadamente de 1 al 2 % de las moléculas del factor VII que fluyen en la sangre, han sido escindidas en la posición Arg 152 para originar la serín proteasa factor VIIa; sin embargo, el centro activo de esta enzima no se expresa eficientemente a menos que el factor VIIa se enlace al FT.28,29 Una vez formado el complejo factor VIIa/FT, este activa al factor X y al factor IX, tanto el factor Xa como el factor IXa formados activan a su vez al factor VIIa, lo que constituye un mecanismo de amplificación. El factor Xa formado, ya sea de forma rápida por el complejo factor VIIa/FT, o de forma más lenta pero continuada por el complejo factor Ixa/VIIIa, activa a la protrombina en presencia de factor Va, fosfolípidos y calcio (complejo protrombinasa). La trombina formada en esta reacción es una serín proteasa potente que acelera de forma dramática el proceso de la coagulación, al modificar proteolíticamente a los factores VIII y V, separa a los fibrinopéptidos A y B de las cadenas a y b del fibrinogéno, lo que permite que este polimerice espontáneamente y forme un polímero de fibrina reforzado por la acción del factor XIIIa, el cual es también producto de la acción de la trombina sobre el factor XIII. El factor XIIIa es una aminotransferasa que une radicales de ácido glutámico y lisina de cadenas g adyacentes, que forma la fibrina insoluble.30
Las teorías originales de la coagulación proponen una serie de reacciones enzimáticas iniciadas por el ensamblaje de las proteínas de la fase de contacto en las superficies cargadas negativamente,31,32 sin embargo, la ausencia de complicaciones hemorrágicas en pacientes con deficiencias de estos factores o el síndrome general-mente moderado en las deficiencias de factor XI, en contraposición con el grave trastorno hemorrágico que producen las deficiencias de los factores IX y VIII y las observaciones experimentales, entre ellas que el complejo factor VIIa/FT, activa tanto al factor X como al factor IX, lo que llevó a plantear en la década de los 80 que el principal evento iniciador in vivo de la coagulación sanguínea era la exposición del FT.33,34 En ese momento quedaba sin esclarecer el papel fisiológico del factor XI, y la variedad de las manifestaciones clínicas que se presentan en los pacientes deficientes de factor XI: algunos pacientes experimentan complicaciones hemorrágicas importantes, mientras otros parecen hemostáticamente normales.35-37 Estas evidencias, conjunta-mente con diferentes observaciones experimentales, han impulsado la búsqueda de un mecanismo alternativo de activación del factor XI independiente de la fase de contacto. A inicios de la década de los 90 Naito y Fujikawa38 demostraron que la trombina activa al factor XI en presencia de sulfato de dextrano; posteriormente se demostró que el factor XI es activado como resultado de la generación de las proteasas formadas durante la interacción de las plaquetas con los factores de la coagulación. Tanto la trombina como el factor Xa pueden activar al factor XI en presencia de plaquetas activadas, pero la activación por la trombina es claramente más importante. Esta reacción puede producirse en ausencia de factor XII, pero requiere la presencia de QAPM y de iones Zn2+.39 Inicialmente se pensó que el complejo XI-QAPM se enlaza a la superficie plaquetaria, sin embargo, se ha demostrado que el QAPM interactúa con el dominio A1 del factor XI, lo que produce una transición conformacional del factor XI que expone sitios de enlace a las plaquetas dentro del dominio A3.40 Más recientemente se determinó que la protrombina y los iones Ca sustituyen al QAPM y a los iones Zn en su función de cofactores en la activación del factor XI.41 Estos mecanismos son independientes de las proteínas de la fase de contacto, obviando la necesidad de estos factores para la hemostasia, lo cual explica la ausencia de defectos en la coagulación en pacientes con deficiencias de estas proteínas y suministra nuevos argumentos para validar la teoría alternativa, también llamada teoría revisada de la coagulación sanguínea, aunque su significación in vivo queda por esclarecer.
En la figura 2 se muestran las interacciones más importantes que ocurren durante la coagulación.

Fig. 2. Interacciones principales durante el mecanismo de la coagulación.
Reacciones de la coagulación sobre superficies celulares
Las superficies celulares constituyen el ambiente natural donde se desarrollan las reacciones de la coagulación sanguínea. Para que se produzca una hemostasia eficaz deben cooperar diferentes tipos celulares. Las plaquetas suministran la superficie más eficiente para la generación de trombina, sin embargo, carecen de FT, y por ello no pueden iniciar la coagulación. Otras células expresan el FT en su superficie y algunas como los monocitos son capaces de ensamblar en su superficie al complejo activador del factor X y al complejo protrombinasa, por lo que para generar trombina de forma eficiente deben participar al menos 2 tipos celulares.42
Usando un sistema celular que semeja las condiciones en las que ocurren las reacciones de la coagulación in vivo, Roberts y otros43 demostraron que el complejo factor VIIa/FT inicia la coagulación activando tanto al factor IX como al factor X en una etapa inicial o de ignición. Los factores Ixa y Xa resultantes tienen funciones muy diferentes en las próximas reacciones.44,45 El factor Xa es necesario para que tenga lugar la activación plaquetaria, y el factor IXa se requiere para que tenga lugar una producción suficiente de trombina. La señal para la activación plaquetaria parece ser la trombina formada por las reacciones que ocurren en la superficie de las células que expresan el FT. Estas pequeñas cantidades de trombina son suficientes también para disociar el factor von Willebrand del factor VIII y activar al factor XI, pero son insuficientes para la formación del coágulo de fibrina estable. Por otra parte, el factor IXa generado por el complejo factor VIIa/FT, actúa como estímulo para la generación significativa de trombina en las plaquetas activadas.46
El factor Xa formado por la acción del complejo factor VIIa/FT no alcanza eficazmente la superficie plaquetaria, debido en parte a su rápida inhibición en la fase fluida por la antitrombina III (ATIII) y por el inhibidor de la vía del FT (IVFT).46,47 Además, las plaquetas deben ser activadas para liberar o unir al factor Va sobre su superficie. Una vez unido a la plaqueta, el factor Xa se encuentra relativamente protegido de la acción inhibitoria de la ATIII. A diferencia del factor Xa, el factor IXa se encuentra mucho más capacitado para viajar a través de la fase fluida y formar complejos en la superficie plaquetaria, pues es inhibido más lentamente por la ATIII y no es neutralizado por el IVFT. Así, el factor IXa es capaz de mantenerse a la espera por más tiempo que el factor Xa, hasta que las plaquetas sean activadas y expresen lugares de unión específicos para el factor IXa.30
La segunda etapa o etapa de propagación se lleva a cabo una vez que las plaquetas son activadas, los factores Va y VIIIa se unen a estas y son responsables del anclaje y orientación de sus respectivas proteasas, lo que permite la expresión de la actividad coagulante. El complejo IXa/VIIIa en la superficie plaquetaria proporciona un suministro continuo de factor Xa asociado con esta superficie, que a su vez posibilita el ensamblaje del complejo protrombinasa, el cual fomenta una generación explosiva de trombina. De esta forma, la única fuente efectiva de factor Xa para el ensamblaje de la protrombinasa plaquetaria la constituye el complejo IXa/VIIIa plaquetario.45,48
En la tercera etapa o de finalización intervienen los diferentes mecanismos inhibitorios de la vía del factor tisular y finalizan la activación de dicha vía.
El mecanismo de la coagulación en la hemofilia A y B
Las deficiencias de factor VIII o de factor IX conducen a una diátesis hemorrágica grave en dependencia del nivel del factor, debido a que cuando el complejo activador del factor X plaquetario no se encuentra formado, las cantidades de factor Xa que alcanzan la superficie plaquetaria son insuficientes para fomentar la generación de trombina en cantidades efectivas. Estas consideraciones han sido validadas por los resultados experimentales, en estudios realizados con sangre y plasma de individuos con hemofilia y con inhibidores de la coagulación. La conclusión más importante obtenida es que la característica distintiva asociada con la etiopatogenia de estas enfermedades es una generación defectuosa de trombina más allá de la formación del coágulo fibrina-plaqueta inicial.6 En estudios extensivos realizados con sangre de individuos con hemofilia A y B se obtuvo que las principales diferencias cuantitativas con respecto al control vistas en la hemofilia A (y reproducidas en la hemofilia B) son: una extensión moderada en la duración de la fase iniciación y una reducción moderada también en la cantidad de fibrinopéptido A acompañada por una supresión en la fase de propagación de la generación de trombina.49 Por lo tanto, en la hemofilia se forma el tapón hemostático primario (plaquetario), pero este se encuentra escasamente estabilizado por fibrina, lo que conduce a un coágulo friable, inestable y fácil de romper.
Inhibidores fisiológicos de la coagulación
Inhibidor de la vía del FT
Este inhibidor regulador de la actividad catalítica del complejo factor VIIA/FT fue purificado por primera vez y clonado a mediados de los años 80, pertenece a la familia de los inhibidores de proteasas tipo Kunitz,50 existe en el plasma humano en 2 formas principales de 40 y 33 kd y en varias formas adicionales de menor peso molecular. En condiciones basales la mayoría del IVFT circula asociado con lipoproteínas. Las plaquetas contienen aproximadamente el 8 % del IVFT y posiblemente este se libera de plaquetas activadas en el sitio de la lesión, lo que contribuye a elevar sustancialmente su concentración local.51 La fuente endógena principal del IVFT es el endotelio vascular, el IVFT es liberado después de la infusión de heparina, lo que eleva sus niveles plasmáticos en varios órdenes de magnitud.52 El IVFT inhibe al factor X directamente, mientras que la inhibición del factor VIIa requiere de la presencia simultánea de factor Xa.
La inhibición del factor VIIa procede en 2 etapas: en la primera se forma el complejo factor Xa-IVFT, que en una segunda etapa se une con el complejo factor VIIa/FT y forma un complejo cuaternario factor Xa-IVFT-VIIa-FT.53 Una hipótesis alternativa plantea la unión directa del IVFT con el complejo factor VIIa-FT-Xa. A concentraciones suprafisiológicas, el IVFT inhibe al complejo factor VIIa-FT en ausencia de factor Xa.54
Sistema de la proteína C
La superficie del endotelio constituye el sitio principal para la activación de la proteína C (PC), la vía de la PC se inicia cuando la trombina se enlaza a la trombomodulina presente en la superficie de las células endoteliales.55 El complejo trombina-trombomodulina es un activador potente de la PC. La activación aumenta cuando esta se enlaza a su receptor en las células endoteliales,56 aunque no todos los complejos de activación involucran al receptor de la PC, ya que los niveles de este son menores en la microcirculación.57
Una vez que se forma la PCa por acción del complejo trombinatrombomodulina, esta inactiva a los factores Va y VIIIa por ruptura proteolítica, limitando así la posterior formación de trombina (fig. 3). La PCa inactiva al factor Va por ruptura en la posición Arg 506, lo que resulta en una rápida pero incompleta pérdida de la actividad y en la posición Arg 306, que ocasiona una inactivación completa.58
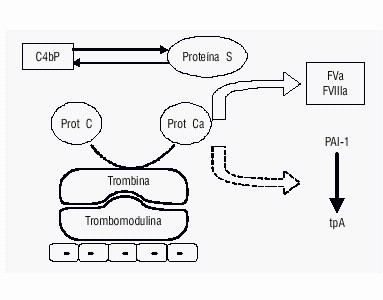
Fig. 3. Acciones de la proteína C en el mecanismo de la coagulación.
La proteína S (PS) actúa cofactor de este proceso: aumenta la afinidad de la PC por la membranas y cambia la especificidad de la ruptura proteolítica del factor Va, ya que acelera la ruptura en la posición Arg306 sin modificación significativa de la velocidad de ruptura en la posición Arg506.59,60 Cuando la PS se enlaza la PCa disminuye en 1 nm la distancia del centro activo de la PCa a la superficie de la membrana; esta disminución es probablemente responsable de la capacidad de la PS de aumentar selectivamente la velocidad de ruptura en la posición Arg306. Otras acciones de la PS son bloquear la capacidad del factor Xa de proteger al factor Va de la inactivación por la Pca y además, en combinación con el factor Va, aumentar la capacidad de la PCa de inactivar al factor VIIIa.61
Antitrombina III
La ATIII es el inhibidor más importante de la coagulación, es un miembro de la familia de inhibidores de serín proteasas conocida como Serpinas.62 La ATIII neutraliza a la trombina por formación de un complejo estequiométrico entre los 2 componentes, a través de la interacción de un residuo Arg del centro reactivo del inhibidor y de la Ser del centro activo de la enzima.63 La formación del complejo ocurre a una velocidad relativamente lenta en ausencia de heparina, sin embargo, cuando el polisacárido está presente, se enlaza con residuos Lys en la ATIII, lo que produce un cambio conformacional en esta proteína, que le permite su unión a la proteasa y se acelera de forma dramática la velocidad de formación del complejo ATIII-trombinaheparina. Una vez formado este complejo, la heparina se disocia del mismo y se une con otras moléculas de ATIII, y el complejo trombina- ATIII es eliminado entonces de la circulación.64
Muchas enzimas del mecanismo de la coagulación son inactivadas por la ATIII: XIIa, XIa, IXa, Xa y VIIa. Este proceso ocurre de forma similar a lo descrito anteriormente para la trombina.63,65 (fig. 4), lo que hace del mecanismo ATIII-heparina la vía principal de neutralización de la mayoría de los factores activados, excepto el factor XIIa, en el cual el mecanismo principal de inhibición lo lleva a cabo el inhibidor del componente C1 del complemento.66
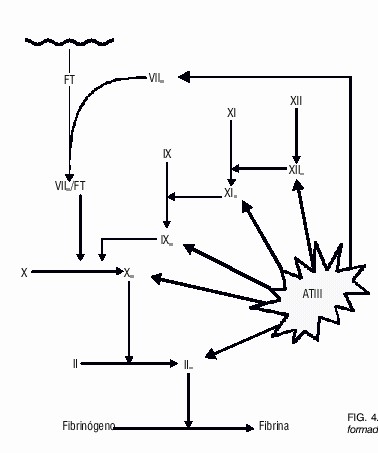
Fig. 4. Inhibición de las serin proteasas formadas durante la coagulación por la ATIII.
In vivo, la acción de la ATIII sobre las proteasas generadas en la coagulación sanguínea es acelerada por el sulfato de heparano y otros glicosaminoglicanos relacionados presentes en la matriz que rodea el endotelio vascular.67
Cofactor II de la Heparina
El cofactor II de la heparina fue aislado por Tollefsen y otros.68 Se enlaza a la heparina con más baja afinidad que la ATIII, pero la velocidad a la cual neutraliza a la trombina se acerca a la de la ATIII en concentraciones saturantes de heparina. La especificidad del cofactor II de la heparina está restringida a la trombina y el sulfato de dermatano. Un mucopolisacárido presente en los vasos sanguíneos puede sustituir a la heparina y acelerar la neutralización de la trombina por el cofactor II de la heparina aproximadamente en 1 000 veces, lo que adquiere mayor importancia, según se plantea, en la microcirculación, donde es mayor la concentración del sulfato de dermatano.69
Summary
At present, it is categorically accepted that the exposure of the tissue factor (TF) is the main initiator event of blood coagulation, which brings about the formation of the factor VIIa/TF complex that activates the factors IX and X on the surfaces of the cells expressing the TF and, thus, the first amounts of thrombin are formed. This thrombin has multiple functions in the haemostatic mechanism, but it is insufficient to attain an efficient haemostasis, which is only possible with the assembly of the prothrombinase complex on the platelet surface. The discovery in the 1990s of the activation of factor XI by thrombin allows to explain the clinical observations made in those patients with deficiency of the coagulation contact phase factors: these patients do not present hemorrhagic complications (excepting the deficit of factor XI). The recent discovery of the activation of precallicrein and factor XI by a cysteine protease located mainly on the surface of the endothelial cells make us think about a new role of these proteins in vivo. Blood coagulation is a closely regulated process and due to its physiological significance it is important the regulation of the tissue factor pathway that limits the initial amounts of thrombin, as well as the regulation of the proteases formed during the mechanism of coagulation by antithrombin III and of the cofactors activated by the protein C system.
Subject headings: BLOOD COAGULATION FACTORS/analysis; PROTHROMBIN/analysis.
Referencias bibliográficas
1. Mann KG, Nesheim M, Church W, Haley P, Krishnaswamy S. Surface dependent reactions of the vitamin K-dependent enzyme complexes. Blood 1990;76:1-6.
2. Stenflo J, Feinlud P, Egan W. Vitamin K dependent modifications of glutamic acid residues in prothrombin. Proc Natl Acad Sci USA 1974;71:2730-3.
3. Furie B, Bouchard B, Furie B. Vitamin K-dependent biosynthesis of gammacarboxziglutamic acid. Blood 1999;93:1798-808.
4. Stenflo J. Contributions of GLA and EGF-like domains to the function of vitamin K dependent factors. Crit Rev Eukaryotic Gene Expression 1999;9:59-88.
5. Mc Donald JF, Shah AM, Schwalbe RA, Kisiel W, Dahlbäck B, Nelsesteuen GL. Comparison of naturally ocurring vitamin K dependent proteins. Biochemistry 1997;36:5120-7.
6. Mann KG. Biochemistry and Physiology of blood coagulation. Thromb Haemost 1999;82:165-74.
7. Schmaier AM, Rojkjaer R, Chiriat-Madar. Activation of the plasma kallikrein/kinin system on cells. A revised hypothesis. Thromb Haemost 1999;82:226-33.
8. Tankersley DL, Finlayson JS. Kinetics of activation and autoactivation of human factor XII. Biochemistry 1984;23:273-9-
9. Dunn JJ, Silverbeg M, Kaplan AP. The cleavage and formation of activated human Hageman factor by autodigestion and by kallicrein. J Biol Chem 1982;257:1779-84.
10. Thompson RE, Mandle R Jr, Kaplan AP. Association of factor XI and high molecular weight kininogen in human plasma. J Clin Invest 1977;60:1376-80.
11. Bouma BN, Griffin JH. Human blood coagulation factor xi. Purification, properties and mechanism of action by activated factor XI. J Biol Chem 1977;253:6432.
12. Gilbert GE, Arena AA. Activation of the factor VIIIa-factor IXa enzyme complex of blood coagulation by membranes containing phosphatidyl-L-serine. J Biol Chem 1996;271:11120-5.
13. Rapaport SI, Proctor RR, Pateh MJ, Yettra M. The mode of inheretance of PTA deficiency: evidence for the existence of a major PTA deficiency and a minor PTA deficiency. Blood 1961;18:149-55.
14. Leiba H, Ramos B, Many A. Hereditary and coagulation studies in ten families with factor XI (plasma thromboplastin antecedent) deficiency. Br J Haematol 1965;11:654-65.
15. Colman RW, Schmaien AH. Contact system: a vascular biology modulator with anticoagulant, profibrinolytic, antiadhesive and proinflamatory attibutes. Blood 1997;90:3819-43.
16. Sidi A, Gelighson V, Jonas P, Many M. Factor XI deficiency detection and management during urologic surgery. J Urol 1978;119:508-30.
17. Hasan AAK, Cines DB, Ngaiza JR, Jaffe E, Schmaier AH. High molecular weight kininogen is exclusively membrane bound on endothelive cells to influence activation of vascular endothelium. Blood 1995;85:3134-43.
18. Zini JM, Schmaier AH, Cines DB. Bradykynin regulates the expression of kininogen binding sites on endothelial cells. Blood 1993;81:2936-46.
19. Motta G, Rojkyaer R, Hasan AAK, Cines DB, Schmaier AH. High molecular weight kininogen regulates prekallikrein assembly and activation of endothelial cells: mechanism for contact activation. Blood 1998;91:516-28.
20. Rojkjaer R, Hasan AAK, Schmaier AH. A novel mechanism for factor XI activation on endothelial cells independent of autoactivation, µ thrombin, or factor XIIa. Blood 1998;92(Suppl 1):40ª.
21. Colman RW, Pixley RA, Najamiennisa S, Yan W-Y, Wang J, Nazar A, McCrac KR. Binding of high molecular weight-kininogen to human endothelial cells is mediated via a site within domain 2 and 3 of urokinasa receptor. J Clin Invest 1997;100:1481-7.
22. Hasan AAK, Krishnan R, Tulinshy A, Srikanth S, Schmaier AH. The mechanism of thrombostatin´s inhibition of thrombin induced platelet activation. Circulation 1998;(Suppl 1): I-800.
23. Bradford HN, De La Cadena RA, Kunapuli SP, Dong JF, Lopez JA, Colman RW. Human kininogens regulate thrombin binding to platelets through GPIb-IX complex. Blood 1997;90:1508-15.
24. Rojkjaer R, Schmaier AH. Activation of the plasma kallikein/kinin system on endothelial cells. Proc Assoc Am Physicians 1999;111:220-7.
25. Camerer E, Kolst A-B, Prydz H. Cell biology of tissue factor the principal initiatior of blood coagulation. Thromb Res 1996;81:1-41.
26. Drake TA, Morrisey JH, Edgington TS. Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am J Pathol 1989;134:1087-7.
27. Morrisey JH, Fakhrai H, Edgington TS. Molecular cloning of the cDNA for tissue factor, the cellular receptor for the initiation of the coagulation protease cascade. Cell 1987;50:129-35.
28. Lawson JH, Butenas S, Mann KG. The evaluation of complex-dependent alterations in the human factor VIIa. J Biol Chem 1992;267:4834-43.
29. Morrisey JH. Macik BG. Neuenschwander PF, Com PC. Quantitation of activated factor VII levels in plasma using a tissue factor mutant selectively deficient in promoting factor VII activation. Blood 1993;81:734-44.
30. Battle J, López Fernández MF. Vía del factor tisular Factor VIIa recombinante. Rev Iberoamer Tromb Hemostasia 1998;11:187-200.
31. Davie EW, Ratnoff OD. Waterfall sequence for intrinsic blood clotting. Science 1964;145:1310-2.
32. Mac Farlane RG. An enzyme cascade in the blood clotting mechanism and its function as a biochemical amplifier. Nature 1964;202:498-9.
33. Zur M, Nemerson Y. Kinetics of factor IX activation via the extrinsic pathway dependence of Km on tissue factor. J Biol Chem 1980;255:5703-7.
34. Marlar RA, Kleiss AJ, Griffin JH. An alternative extrinsic pathway of human blood coagulation. Blood 1982;60:1353-8.
35. Ragni MV, Sinha D, Seaman F, Lewis JH, Spero JA, Walsh PN. Comparison of bleeding tendency factor XI coagulante activity, and factor XI antigen in 25 factor XI.deficient kindres. Blood 1985;65:719-24.
36. Bolton-Maggs PH, Young Wan-Yin B, McCraw AH, Slack J, Kernoff PB. Inheretance and bleeding in factor XI deficiency. Br J Haematol 1988;69:521-8.
37. Asakai R, Chung DW, Davie EW, Selighsohn U. Factor XI deficiency in Ashkenazi Jews in Israel. N Eng J Med 1991;335:153-8.
38. Naito K, Fujikawa K. Activation of human blood coagulation factor XI independent of factor XII. Factor XI is activated by thrombin in the presence of negatively charged sufaces. J Biol Chem 1991;266:7353-8.
39. Oliver JA, Monroe DM, Hoffman M, Roberts HR. Factor XI increases thrombin generation in a cell based system in the absence of factor XII. Blood 1996;(Suppl 1):1863 (abstract).
40. Baglia FA, Jameson BA, Walsh PN. Identification an characterization of a binding site for platelets in the Apple 3 domain of coagulation factor XI. J Biol Chem 1995;270:6734-40.
41. Baglia FA, Walsh PN. Prothrombin is a cofactor for the binding of factor XI to the platelet surface and for platelet mediated factor XI activation by thrombin. Biochemistry 1998;27:2271-1.
42. Hoffman M, Monroe DM; Roberts HR. Cellular interactions in hemostasis. Haemostasis 1996;(Suppl 1):12-6.
43. Roberts HR, Monroe DM, Oliver JA, Chang J-Y, Hoffman M. New concepts of blood coagulation. Haemophilia 1998;4:331-4.
44. Monroe DM, Roberts HR, Hoffman M. Platelet procoagulant complex assembly in a tissue factor initiated system. Br J Haematol 1994;88:364-71.
45. Hoffman M, Monroe DM, Oliver JA, Roberts HR. Factors IXa and Xa play distintict roles in tissue dependent initiation of coagulation. Blood 1995;86:1794-1801.
46. Monroe DM, Hoffman M, Roberts HR. Transmission of a procoagulant signal from tissue factor bearing cells to platelets. Blood Coag Fibrin 1996;7:459-65.
47. Rapaport SI. Inhibition of factor VIIa-tissue factor induced blood coagulation with particular emphasis on a factor Xa-dependent inhibitory mechanism. Blood 1989;73:359-64-
48. Rapaport SI, Rao LVM. The tissue factor pathway: how it has become a prima ballerina. Thromb Haemost 1995;74:7-17.
49. Cawthern KM, van´t Veer C, Locke JB, DiLorenzo ME, Brands RF, Mann KG. Blood coagulation in hemophilia A and hemophilia C. Blood 1998;91:4581-92.
50. Wun TC, Kretzmer KK, Girard TJ, Miletich JP, Broze GJ Jr. Cloning and characterization of cDNA coding for the lipoprotein associated coagulation inhibitor shows that it consists of three tandem Kunitz-type inhibitor domains. J Biol Chem 1988;263:6001-4.
51. Novotny WF, Girard TJ, Miletich JP, Borze GJ Jr. Platelet secrete a coagulation inhibitor functionally and antigenically similar to the lipoprotein associated coagulation inhibitor. Blood 1988;72:2020-5.
52. Sandset PM, Abilgaard U, Larsen ML. Feparin induces release of extrinsic coagulation pathway inhibitor (EPI). Thromb Res 1988;50:803-8.
53. Broze GJ, Warren LA, Novothy WF. Higuchi DA, Girard H, Miletich JP. The liporotein associated coagulation inhibitor that inhibits factor Xa: Insight into its possible mechanism of action. Blood 1988;71:335-43.
54. Callander NS, Rao LVM, Nordfang O, Sandset PM, Warn-Cramer B, Rapaport SI. Mechanisms of binding of recombinant extrinsic pathway inhibitor (rEPI) to cultured cell surfaces. J Biol Chem 1992;267:876-82.
55. Esmon CT. The roles of protein C and thrombomodulin in the regulation of blood coagulation. J Biol Chem 1989:264:4743-6.
56. Stearns-Kurosawa DJ, Kurosawa S, Mollica JS, Ferrell GL, Esmon CT. The endothelial cell protein C receptor augments protein C activation by the thrombin-thrombomodulin complex. Proc Natl Acad Sci USA 1996;93:10212-6.
57. Laszick Z, Metro A, Taylor FBJr, Ferrell GL, Esmon CT. Human protein C receptor is present primarily on endothelium of large blood vessels: implications for the control on the protein C pathway. Circulation 1997:96:3633-40.
58. Egan JO, Kalafatis M, Mann KG. The effect at Arg 306 Ala and Arg 504 GLn substitutions in the inactivation of recombinant human factor Va by activated protein C and protein S. Protein Sci 1997;6:2016-7.
59. Walker FJ. Regulation of activated protein C by a new protein. A role of bovine protein S. J Biol Chem 1980;255:5521-4.
60. Lu D, Kalafatis M, Mann KG, Long GL. Comparison of activated protein C/protein S-mediated inactivation of human factor VIII and factor V. Blood 1996;87:4708-17.
61. Shen L, Dahlbäck B. Factor V and protein S as synergistic cofactors to activated protein C in degradation of factor VIIIa. J Biol Chem 1994;369:18735-8.
62. Carrell RV, Chirstey PB, Boswell DR. Serpins: antithrombin and other inhibitors of coagulation and fibrinolysis in Vers. Traete M. Vermylen J, Lijnen HR, et al. (eds). Thrombosis and Hemostasis, Brussels: Leuven University Press, 1987:1-15.
63. Rosenberg RD, Damus PS. The purification and mechanism of action of human antithrombin-heparin cofactor. J Biol Chem 1973;248:6490-6505.
64. Shore JD, Olson ST, Craig PA. Kinetics of heparin action. Ann NY Acad Sci 1989;556:75-80.
65. Stevens AW, Siddqui A, Hirs CHW. Expression of functionally active human antihtrombin III. Proc Natl Acad Sci USA 1987;84:3886-90.
66. Bjork L, Danielsson A. Antitrombin and related inhibitors of coagulation proteinases. En: Barrett AJ, Salvesen GS (eds). Proteinase inhibitors. Amsterdan: Elsevier, 1986:489-513.
67. Marcum JA, Rosenberg RD. Heparanlike molecular with anticoagulant activity are synthesized by culture endothelial cells. Biochem Res Comun 1985;126:365-72.
68. Tollefsen DM, Blank MK. Detection of a new heparin dependent inhibitor of thrombin in human plasma. J Clin Invest 1981;68:589-96.
69. Comp PC. Overview of the hypercoagulable states. Sem Thromb Hemost 1990;16:158-611.
Recibido: 22 de diciembre del 2000. Aprobado: 10 de enero del 2001.
Lic. Alina Díaz Concepción. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800, Ciudad de La Habana, Cuba. Telef: (537)578268. Fax: (537)338979. mailto:ihidir@hemato.sld.cu

