










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter v.19 n.2-3 Ciudad de la Habana Mayo-dic. 2003
Síndrome hemolítico urémico
Dr. Juan Carlos Jaime Fagundo,1 Dr. Yosniel Delgado Giniebra,2 Dra. Dunia Castillo González,1 Dra. Valia Pavón Morán,1 Dra. Anadely Gámez Pérez 3 y Dr. Luis A. Sánchez Mallo 4
Resumen
Se realiza una revisión de los aspectos generales del síndrome hemolítico urémico (SHU): epidemiología, patogenia, manifestaciones clínicas y tratamiento. El SHU es el resultado de la acción de numerosos factores etiológicos y patogénicos, causado principalmente por una verotoxina producida por diferentes cepas de Escherichia coli. En esta entidad se produce un daño endotelial que genera una serie de fenómenos tales como: adhesión, agregación plaquetaria y depósitos de fibrina, que llevan a la formación de trombos en la microcirculación: la microangiopatía trombótica. El SHU se caracteriza por la combinación de insuficiencia renal aguda, trombocitopenia y anemia hemolítica microangiopática. Las diarreas y las infecciones del tracto respiratorio superior son los factores precipitantes más comunes. La plasmaféresis y la diálisis son actualmente el tratamiento de elección.
DeCS: SINDROME HEMOLITICO- UREMICO/ epidemiología; SINDROME HEMOLITICO-UREMICO/ etiología; SINDROME HEMOLITICO-UREMICO/ quimioterapia; ESCHERICHIA COLI; INSUFICIENCIA RENAL AGUDA; TROMBOCITOPENIA; ANEMIA HEMOLÍTICA; NIÑO.
Microangiopatía trombótica (MAT) es un término anátomo-patológico que describe la existencia de trombosis en los pequeños vasos del organismo. Algunos autores adoptan esta denominación para referirse en su conjunto a 2 enfermedades: el síndrome hemolítico urémico (SHU) y la púrpura trombocitopénica trombótica (PTT), los cuales comparten muchas características comunes.1
Gasser y colaboradores 2 fueron los que describieron el SHU en 1955. Treinta años antes, Moschowits 3 reportó el caso de una adolescente que falleció de microangiopatía trombótica y anemia. Este trastorno, inicialmente llamado síndrome de Moschowits y después conocido como PTT, es muy similar al SHU. Este último constituye la causa más frecuente de insuficiencia renal aguda (IRA) en el niño.
Aunque aproximadamente en el 90 % de los niños el cuadro es precedido por un episodio diarreico (SHU clásico), esta entidad puede ser secundaria a gran variedad de situaciones.
Definición
El SHU es un cuadro caracterizado por la combinación de insuficiencia renal aguda, trombocitopenia y anemia hemolítica microangiopática. Afecta tanto a los niños como a los adultos y está causado, en la mayoría de los casos, por cepas de Escherichia coli productoras de verotoxinas (E. coli enterohemorrágica, ECEH). Es la más frecuente la del serotipo 0157: H7. 1,2
Clasificación del SHU
En la actualidad existen varias clasificaciones, pero la de Kaplan es considerada una de las más prácticas, la cual se muestra a continuación.
Clasificación de Kaplan- Idiopático
- Secundario a:
a) Infecciones asociadas con SHU: E.coli 0157:H7, Shigella dysenteriae tipo I, Streptococus pneumoniae.
b) Infecciones circunstanciales: Salmonella tiphy, Campylobacter fetus jejuni, Yersinia pseudotuberculosa, Bacterioides , virus Portillo, Cocksackie virus, ECHO virus, influenza, Epstein Barr , rotavirus ,VIH, Microtabiotes, etc.
c) Forma genética: herencia autosómica recesiva y formas dominantes.
d) Asociado con drogas: anticonceptivos orales, ciclosporina A, mitomicina.
e) Durante el embarazo, postrasplante de médula y riñón, asociado con glomerulopatías y procesos malignos.
f) Formas recurrentes esporádicas autosómicas recesivas o dominantes.
La clasificación de Neil,4 aunque similar a la de Kaplan, se considera la más compleja, debido a que presenta una división etiológica de la enfermedad en relación con las diferentes formas en que se presenta:
- Idiopática.
- Debido a:
a) Causas infecciosas:
- Asociada con diarreas (E. coli 0157H7).
- Asociada con Shigella dysenteryae tipo 1.
- Asociada con neuraminidasa (Streptococcus pneumoniae).
- Asociada con otras infecciones circunstanciales (Salmonella typhi, Campylobacter fetus jejuni, Yersinia pseudotuberculosis, bacterioides, virus Portillo, Cocksackie, ECHO virus, influenza, Epstein Barr, rotavirus, HVI, microtatobiotos).
b) Causas no infecciosas (esporádico):
- Familiar (herencia autosómica recesiva y formas dominantes).
- Tumores.
- Drogas (contraceptivos orales, ciclosporina A, mitomicina C, FK 506, OKT3, metronidazol, penicilina, cisplatino, daunorubicina, arabinósido de citosina, deoxycoformicina, ticlopidina, quinina).
- Embarazo.
- Enfermedades sistémicas.
- Trasplantes (médula ósea, riñón).
- Glomerulonefritis.
- Formas recurrentes esporádicas autosómica recesiva o dominante.
Además de las causas de SHU descritas en esta clasificación, se reportan otras como la infección por Entamoeba histolítica y a la administración de algunas vacunas como la DPT, antipoliomielitis, sarampión, rubéola y parotiditis.
También se ha descrito una variedad de este síndrome consecuente con errores congénitos del metabolismo de la cianocobalamina y a la inhalación de crack-cocaína, dado la vasoconstricción que produce esta sustancia junto al daño endotelial que establece y a sus efectos procoagulantes.5
Epidemiología
Aunque siempre se relaciona la presencia de SHU con el antecedente de diarreas tipo invasivas, no es así en todos los casos, pues existe una división importante en SHU típico o D+, que presenta como antecedente enfermedades diarreicas agudas, y el atípico o D-, que no tiene este antecedente (tabla 1).
Tabla1. Diferencias entre el síndrome hemolítico urémico típico (SHU D+) y el SHU atípico (SHU D-)
| Características | SHU D+ | SHU D- |
| Antecedentes patológicos familiares | No | Si |
| Edad | < 1 año | > 5 años |
| Variedad estacional | Sí | No |
| Pródromos | Diarrea | No diarrea |
| Hipertensión endocraneana | Moderada | Severa |
| Complicaciones | Poco comunes | Comunes |
| Histología | Microangiopatía glomerular | Arteriopatía |
| Recurrencia | Rara | Común |
| Frecuencia | 90 % de los casos | 10 % de los casos |
| Evolución | Buena | Mala |
| Factor vW disfuncional | Presente | Ausente |
| Alteración factor H | No | Sí |
La ECEH O157: H7, que es el agente causal de la colitis hemorrágica, produce una potente toxina conocida como Shiga-like toxin, llamada así por su semejanza con la producida por la Shigella dysenteriae o Verotoxina, como también se le conoce (figura).
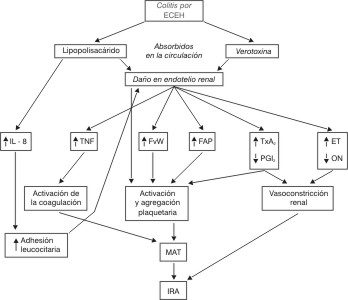
Fig. Cascada patogénica abreviada del SHU posdiarreico.
ECEH: E. coli enterohemorrágica; IL-8: interleucina 8; TNF: factor de necrosis tumoral; FvW: factor von Willebrand; FAP: factor activador plaquetario; TxA2: tromboxano A2 ; PGI2 : prostaciclina; ET: endotelio; ON: óxido nítrico; MAT: microangiopatía trombótica; IRA: insuficiencia renal aguda.
Basados en cultivos de heces fecales, hay evidencias que relacionan a la infección por la ECEH con el clásico SHU posdiarreico debido a la presencia de citotoxina fecal o de anticuerpos en el paciente, aunque una gran variedad de organismos han sido implicados en la patogénesis del SHU.
En cuanto a las razas, la mayoría de los estudios muestran que los niños blancos son más susceptibles que los negros.6
Actualmente se conoce que puede aparecer en cualquier parte del mundo y que su frecuencia está aumentando. Existen zonas endémicas en Argentina con alta incidencia, debido posiblemente a la elevada frecuencia de enfermedades diarreicas agudas producidas por toxinas tipo Shiga demostradas mediante serología, neutralización de citotoxinas fecales, cultivo de heces e hibridación del DNA. 6 Existen otras zonas endémicas en África meridional, el oeste de los Estados Unidos de Norteamérica, Holanda, etc. Dentro de las zonas endémicas, se han notificado verdaderos brotes epidémicos y con relativa frecuencia se describen casos esporádicos.
En general, el SHU afecta preferentemente a los lactantes y a los niños menores de 5 años, y es mucho menos frecuente en adultos. No obstante, la edad media de las distintas series publicadas difiere considerablemente de unos países a otros. En África meridional, la edad media es de 8,5 meses, en Argentina de 9,5 meses, 6 en Holanda de 23 meses y en las distintas zonas de los Estados Unidos, oscila entre 3 y 4,5 años. En relación con el sexo, algunos han descrito una mayor incidencia en el femenino. 7
Reservorios y vectores de la infección:La ECEH habita de forma asintomática el intestino del ganado vacuno; especialmente en terneras y novillas.
Debido a que esta cepa bacteriana habita el intestino de estos animales, la mayoría de los brotes están relacionados con la contaminación de productos cárnicos. La hamburguesa es el vector que favorece más de la mitad de los casos epidémicos reportados. El agua y otros productos contaminados con las heces del ganado vacuno, son vectores adicionales de la infección, así como la leche y el yogurt sin pasteurizar. Su vía de transmisión se ha demostrado que es de persona a persona.
Etiología
El SHU/PTT puede corresponder a etiologías muy diversas, entre las cuales pueden citarse:
- La forma clásica o idiopática, frecuente en niños y que puede asimismo presentarse en adultos.
- Las formas posinfecciosas, descritas tras infecciones por Shigella, Salmonella, Escherichia coli, Klebsiella pneumoniae, Pseudomonas, Estreptococos, Yersinia y varios tipos de virus (Coxsackie, ECHO, adenovirus, etc.), incluido el virus de la inmunodeficiencia adquirida (VIH-1).
- Las formas familiares hereditarias y recurrentes.
- Las formas de patogenia inmunológica, que pueden cursar con hipocomplementemia, depósitos glomerulares de C3 y de forma excepcional, presencia de C3 nephritic factor.
- Las formas asociadas con complicaciones del embarazo, el postparto o el uso de anovulatorios.
- Las formas asociadas con diversas enfermedades sistémicas, como el lupus eritematoso sistémico, la esclerodermia, la nefropatía por radiación, el trasplante renal y la hipertensión arterial esencial y la maligna.
- Formas asociadas con fármacos. Además de los anovulatorios, el SHU/PTT se ha descrito con el uso de la mitomicina C, ciclosporina A y antiinflamatorios no esteroides (fenil-butazona, diclofenaco y quinina).
- Formas asociadas con enfermedades glomerulares. El SHU/PTT puede complicar el curso de varias glomerulopatías: síndrome nefrótico por lesiones mínimas, nefropatía membranosa, glomerulonefritis mesangiocapilar y nefropatía IgA.
Anatomía patológica
Las alteraciones en el SHU se caracterizan por la presencia de un material hialino (eosinofílico) en la luz de pequeñas arteriolas y capilares, inicialmente compuesto por plaquetas con algunos depósitos de fibrina. También son frecuentes los depósitos subendoteliales hialinos. El estudio inmunohistoquímico del trombo ha demostrado la presencia de factor von Willebrand (FvW) con una pequeña cantidad de fibrinógeno-fibrina, lo contrario de lo que sucede en las lesiones trombóticas de la coagulación intravascular diseminada (CID), hallazgo que confirma la teoría de agregación plaquetaria mediada por multímeros de FvW.
Las arteriolas renales presentan un engrosamiento de la íntima e hipertrofia de las células de la capa muscular. La presencia de trombos fibrinoides intraluminales es muy llamativa, y a menudo se acompaña de necrosis de la pared vascular. Las arterias interlobulillares también se encuentran afectadas, y presentan un engrosamiento de la íntima, de aspecto mucinoso, que contribuye a reducir el calibre de la luz vascular. En los glomérulos puede observarse un engrosamiento uniforme de las paredes capilares, con posible formación de doble contorno, y a veces trombosis y focos de necrosis. 8,9
La inmunofluorescencia demuestra la presencia de fibrinógeno en las paredes y luz vascular, y en ocasiones en los glomérulos, donde también puede haber depósitos de complemento.
En la microscopía electrónica, los glomérulos se caracterizan por el ensanchamiento del espacio subendotelial, debido a la acumulación de un material poco denso y finamente granular entre el endotelio y la membrana basal glomerular.
En estas condiciones, la síntesis de una nueva lámina basal adosada al endotelio sería responsable del aspecto en doble contorno observado al microscopio óptico.
En los casos más graves de SHU, y en especial, los relacionados con trastornos obstétricos, pueden aparecer áreas de necrosis cortical. 10,11
Patogenia y fisiopatología
A partir de los datos iniciales de Karmali,12 las investigaciones patogénicas realizadas en muchos países señalan que la E. coli productora de toxina citopática, es la causa de la enfermedad. La asociación entre E. coli productora de verotoxina y el SHU, está mediada por la producción de una citotoxina similar a la exotoxina que produce la Shigella dysenteriae tipo 1; por esa razón, también es llamada toxina tipo Shiga (SLT, del inglés Shiga like-toxin).12
En 1977 se aislaron algunas cepas de E. coli, y se encontraron toxinas con las mismas características de la toxina Shiga. 13 El serotipo E. coli 0157:H7 demostró ser uno de los mayores productores de verotoxinas 1 y 2, homólogas respectivamente a las producidas por la Shigella.13
En 1983, Karmali 12 fue el primero en demostrar la función etiológica de las verotoxinas producidas por la E. coli en el SHU.
El trastorno subyacente básico consiste en una lesión del endotelio vascular, que provoca una activación local de la coagulación, con la formación de trombosis en los vasos pequeños. 9
Todos los agentes que pueden inducir SHU/PTT son capaces también de lesionar el endotelio vascular. Así, las endotoxinas bacterianas resultan lesivas para el endotelio vascular. La ciclosporina A y la mitomicina C, fármacos relacionados con el SHU/PTT, pueden causar una lesión directa de la célula endotelial. Además, las células endoteliales sintetizan y liberan diversos factores que interfieren en los procesos hemostáticos y trombóticos.
La E. coli 0157: H7 productor de la verotoxina o la Shigella, se ingiere con los alimentos contaminados poco cocidos (carne, leche no pasteurizada, etc.), colonizan el intestino grueso y se adhieren a las células epiteliales de la mucosa del colon. Después de invadir y destruir dichas células, el tejido subyacente y su vascularización, se produce la diarrea, que generalmente es hemorrágica.
Para que se produzca este mecanismo, la subunidad B de la verotoxina se une con un receptor de membrana que es un glicolípido neutral: el globotriaosylceramide (Gb 3), capaz de reconocerla y penetrar dentro de las células por endocitosis.13 La subunidad A hidroliza un residuo de adenina de la unidad ribosomal 60s, y como consecuencia, las toxinas destruyen la maquinaria proteica de la célula susceptible.
Sin embargo, se han encontrado niveles bajos de Gb3 en algunos pacientes que desarrollan SHU en comparación con controles sanos y con niños que presentan diarrea asociada con bacterias productoras de verotoxinas. Como consecuencia del daño celular que estas producen, se libera FvW, el cual se une con los receptores situados en la membrana de las plaquetas, lo que da lugar a la agregación plaquetaria, a la formación de microtrombos y a la aparición de trombocitopenia.13
El efecto dañino de las toxinas bacterianas sobre las células endoteliales renales y de otros órganos puede ser potenciado por otras sustancias, como los lipopolisacáridos (un componente de las Shigella dysenteriae y de la E. coli 0157: H7), las interleucinas y el factor de necrosis tumoral (liberado por los monocitos, macrófagos y posiblemente por las células mesangiales expuestas a endotoxinas). Las proteasas liberadas por los neutrófilos contribuyen también al daño de las células endoteliales y explican la relación entre la intensidad de la neutrofilia y un mal pronóstico.13
Se ha descrito una disminución de la función de PGI2 secundaria al daño de las células endoteliales, lo que rompe el equilibrio existente entre la formación de PGI2 y TXA2, y favorece la agregación plaquetaria.14 También hay una liberación por parte de las células endoteliales del inhibidor del activador del plasminógeno tipo I, lo cual impide la dilución de los microtrombos formados.
El daño de las células endoteliales glomerulares produce una disminución de la luz glomerular, que es favorecida por la liberación de citocinas de potente acción vasoconstrictora, lo cual aumenta la resistencia vascular y disminuye el flujo sanguíneo renal, que trae como resultado la disminución de dicho flujo, y lleva de esta forma a la insuficiencia renal.
Por otro lado, la anemia hemolítica es secundaria a la disminución de la luz de los pequeños vasos. De esta manera, cuando los eritrocitos pasan por estos vasos con la luz disminuida, se dañan y se fragmentan, y adquieren forma abigarrada (fragmentocitos). Sin embargo, también se describen alteraciones de la membrana eritrocitaria que los llevan a unirse con los multímeros de FvW y trombospondina, de tal forma que se adhieren a la pared vascular y se rompen por la presión alta existente debido a la disminución de la luz capilar.
Existe en la actualidad evidencia que la forma no diarreica del SHU(SHU D-), está asociada con alteraciones y mutaciones de la proteína plasmática multifuncional y multidominio del factor H, 15 y varios estudios han mostrado la clara asociación de niveles bajos de complemento con mutaciones del gen del factor H en el SHU D-. 16,17
El factor H, entre otras funciones, actúa como un regulador temprano de la cascada del complemento. Este factor determina el destino de las moléculas de C3b recién generadas, y actúa como cofactor del factor I en la degradación de C3b, regulando la estabilidad y formación de la convertasa de la vía alternativa del complemento C3bBb. 17
Ante una agresión endotelial de cualquier origen, la activación de plaquetas y leucocitos contribuye a promover la formación de esta convertasa, la cual a su vez lleva al clivaje del tercer componente del sistema de complemento (C3), a su forma activa C3b, disponible para reaccionar con cualquier superficie celular.
En los últimos 20 años, se han descrito cerca de 140 casos familiares de SHU/PTT en 70 familias, donde se han encontrado niveles séricos reducidos del tercer componente del sistema de complemento (C3), lo que reafirma el hecho que la enfermedad es una anomalía congénita hereditaria.15
Síndrome hemolítico urémico D- o atípicoEl SHU D- (asociado con una infección por neumococo), se caracteriza por anemia hemolítica con prueba de Coombs positiva. La neuroaminidasa que produce, rompe el ácido siálico del enterocito, las plaquetas y membrana endotelial, desdobla el ácido neuramínico de las células y deja al descubierto el antígeno de Thomson-Friedereich (T-F), que al exponerse a los anticuerpos IgM que pueden estar presentes en el plasma, producen poliaglutinación con hemólisis, trombosis intravascular y lesión vascular. Esto se agrava con el uso de productos de la sangre que tienen anticuerpos anti T-F, porque produce un proceso de autohemólisis de los hematíes con el empeoramiento del enfermo.
Esta variante de SHU, aunque poco frecuente, debe ser conocida y diagnosticada precozmente, porque en ocasiones, los anticuerpos IgM antiantígeno T-F se infunden al paciente con los derivados de la sangre, y favorecen todavía más la aglutinación. 18 Esta es probablemente la causa de que el índice de mortalidad y morbilidad de esta forma del SHU sea elevado. El SHU asociado con neumococo debe sospecharse cuando ante un paciente con aspecto tóxico, se detectan uno o más de los siguientes factores: neumonía, prueba de Coombs positiva, anemia hemolítica sin respuesta reticulocitaria, o cuando hay dificultades en realizar las pruebas cruzadas de los grupos ABO. Existen otras formas de SHU D- asociadas con una herencia autosómica recesiva y presencia de hipocomplementemia con bajos niveles de C3, que pueden aparecer en generaciones de una misma familia.
Basados en criterios clínicos de familias afectadas, se han establecido varios grupos de SHU con incidencia familiar:
Primer grupo: incluye a los casos donde los familiares afectados desarrollan un SHU en un intervalo de días o semanas. Se trata de casos que normalmente ocurren en áreas endémicas o en brotes epidémicos, que tienen una fase prodrómica diarreica, que no suelen tener recurrencia y con un buen pronóstico. Estos pacientes parecen tener formas adquiridas-infecciosas del SHU.
Segundo grupo: incluye a los casos cuyos familiares desarrollan esta entidad, y son mayores de 1 año. Generalmente no tienen la fase prodrómica, recurren con frecuencia y el pronóstico es pobre. Tienen una forma autosómica recesiva del SHU.
Tercer grupo: incluye a los casos que se transmiten de forma autosómica dominante. Esta forma es la más frecuente en el adulto y tienen índice de mortalidad extremadamente alto. En algunos de estos pacientes, el SHU es desencadenado por el embarazo y la administración de anticonceptivos orales.
Alteraciones de la coagulación en el SHU
Se describen 4 mecanismos fundamentales, que explican las alteraciones a este nivel:
- Daño endotelial.
- Activación plaquetaria.
- Patrón alterado de multímeros del factor von Willebrand.
- Anomalías del sistema fibrinolítico.
El endotelio vascular normal es una interfase metabólicamente activa entre la sangre y los tejidos extravasculares. Su superficie interna es antitrombótica y se involucra en la regulación de la función plaquetaria y la coagulación sanguínea.
El endotelio es el sitio principal de producción de sustancias anticoagulantes como prostaciclina y ácido nítrico. Muchos esfuerzos experimentales han tratado de demostrar que el concepto de injuria endotelial lleva a una pérdida de las propiedades anticoagulantes y antitrombóticas, y contribuyen a la patogénesis de la MAT.9,19 Los estudios histopatológicos de los capilares glomerulares del riñón muestran engrosamiento endotelial y deterioro de la membrana basal en el SHU. 19
Hay evidencias que demuestran que tanto en el SHU como en la PTT el evento primario que lleva al MAT es el daño endotelial, y la activación plaquetaria es solo secundaria a este fenómeno. 19
Las alteraciones de la función procoagulante y protrombótica tienen lugar cuando las células endoteliales son expuestas a ciertas citoquinas o lipopolisacáridos en el SHU y la PTT.
Vale considerar que citoquinas como el FNT-a y la IL-Ib actúan sinérgicamete con los lipopolisacáridos y regulan la sobre expresión de receptores Gb3 en las superficies celulares, incrementando los sitios de unión específica para la Shiga toxina-1 de 1 a 100 veces, lo cual aumenta considerablemente la susceptibilidad de las células endoteliales a las Shiga-toxinas-1.
Las Shiga-toxinas-1 modulan la interacción leucocito-endotelio incrementando la adhesión leucocitaria 20 y la sobre expresión de proteínas adhesivas en la superficie endotelial.
El número de neutrófilos está elevado en el SHU, lo cual es un factor predictivo para la evolución de estos pacientes.20 Estas células en los niños con SHU en fase aguda, se adhieren a las células endoteliales más que los neutrófilos normales, e inducen un daño endotelial al degradar la fibronectina endotelial, debido a la liberación de proteasas neutrofílicas. Esta activación leucocitaria se produce por la liberación en el SHU de sustancias como la alfa 1 antitripsina y la IL-8, esta última es un potente activador de los neutrófilos, y aumenta considerablemente en la fase aguda del SHU. 21
Activación plaquetaria
Está bien aceptado que en niños con SHU y PTT se demuestra la presencia de hemólisis microangiopática y la formación de trombos plaquetarios sin activación de la coagulación sistémica, lo cual es sugestivo de una activación plaquetaria primaria o secundaria.
Fong y Kaplan 22 han reportado que las plaquetas de los niños con SHU presentan disminución de la agregación. Los niveles de P-selectina están significativamente elevados en pacientes con PTT, comparados con los controles, lo que indica activación plaquetaria primaria en esta enfermedad. Además, se demostró una alta concentración urinaria de factor activador plaquetario (PAF) en 10 niños durante la fase aguda de la enfermedad, lo que demuestra la intensificación de la función plaquetaria. 23
Quizás el más importante de estos factores sea la prostaciclina (PGI2), una prostaglandina que tiene propiedades vasodilatadoras y actúa como un potente inhibidor endógeno de la agregación plaquetaria. La PGI 2 inhibe la agregación plaquetaria al aumentar los niveles de AMP cíclico en las plaquetas e inhibe la acción del tromboxano A2, que tiene potentes efectos vasoconstrictores y promueve la agregación de las plaquetas.14
En condiciones normales, existe un equilibrio entre la PGI2 del endotelio vascular y el tromboxano A2 de las plaquetas. Cualquier factor que altere este equilibrio altera la interacción entre las plaquetas y el endotelio vascular. Una de las teorías planteadas para explicar la patogenia del SHU/PTT, que en los últimos años ha tenido mayor difusión, postula que en estos pacientes se halla disminuida la capacidad para sintetizar PGI2.14 Además, los enfermos con SHU/PTT pueden carecer de un factor presente en el plasma normal que estimularía la secreción de esta prostaglandina. Dicha hipótesis adquirió mayor relevancia al demostrarse que se conseguían remisiones del SHU tras la perfusión de plasma fresco. Puede asimismo existir un desequilibrio entre los diversos factores vasoactivos derivados del endotelio (óxido nítrico, endotelina, entre otros). 14
Patrón alterado de multímeros del factor von WillebrandEl endotelio vascular es la fuente de factor von Willebrand circulante, el cual es muy importante para una adhesión plaquetaria eficiente. Los multímeros de factor vW son marcadores importantes del daño endotelial. Ellos están formados por monómeros que se unen por puentes disulfuros y se almacenan dentro de los cuerpos de Weibel-Palade en las células endoteliales y en los gránulos a de los megacariocitos.
Los niveles de factor vW están aumentados en la fase aguda en los pacientes con SHU D+ y se encuentran disminuidos en la fase convaleciente del SHU. 24
La tendencia protrombótica en el SHU puede evidenciarse en parte por la unión más eficiente de los grandes multímeros del factor vW con los complejos glicoproteicos GP Ib-IX y GP Ib-IIIa. La aparición de estos grandes multímeros está dada por la deficiencia de una proteasa escindidora del factor vW. Una deficiencia constitucional de esta proteasa se ha encontrado en casos familiares de PTT, mientras que la forma adquirida es causada por la presencia de anticuerpos inhibidores. 25 En los pacientes con SHU se ha establecido una actividad normal de esta proteasa escindidora. 25
En niños con SHU, los multímeros del factor vW de alto peso molecular están elevados durante la fase aguda y en las recaídas, lo que indica persistencia del daño endotelial en los pacientes con SHU recurrente. Otros autores no han encontrado diferencias entre el antígeno del factor vW entre pacientes con SHU D+ y SHU D-. 24
La variedad de patrones de los multímeros de factor vW no está completamente claro. Se han realizado estudios que utilizan anticuerpos monoclonales contra epítopes específicos de este factor que interactúa con el receptor GP Ib-IX, lo que indica que el factor vW está involucrado en el aumento de la retención plaquetaria en la PTT recurrente. 25 Esto ha sugerido que la heterogeneidad de multímeros de factor vW es removida por agregación con plaquetas en la fase aguda de la enfermedad y por lo tanto, no siempre puede detectarse en la circulación. Esos multímeros pueden ser responsables de la agregación plaquetaria y la adhesión.
Queda por discutir la posibilidad de la trombogénesis a través de la hemólisis, hipótesis que se asume porque las sustancias liberadas por los eritrocitos, por ejemplo el ADP, pueden causar agregación plaquetaria estimulando la formación de coágulos. Se ha demostrado que el ADP bajo condiciones de alto flujo puede causar agregación plaquetaria. Sin embargo, pacientes con hemólisis causadas por otras enfermedades como la eritroblastosis o esferocitos, no tienen trombosis. 25
Involucración del sistema fibrinolíticoEl endotelio vascular es fuente del activador tisular del plasminógeno (t-PA) y secreta inhibidor del activador del plasminógeno tipo I (PAI-1). El daño endotelial presente en el SHU y PTT puede ser responsable de la disminución de la fibrinólisis. Van Hinsberg y colaboradores han estudiado extensamente el sistema fibrinolítico en el SHU. 26 Los inhibidores de la fibrinólisis han sido identificados en niños con SHU, y su remoción por diálisis peritoneal ha sido asociada con una mejoría de la función renal.
El concepto de que una fibrinólisis anormal causa trombosis de la micro vasculatura, es interesante, y potencialmente tiene consecuencias terapéuticas, como por ejemplo el uso exitoso de t-PA en pacientes con SHU.
Estudios in vitro han demostrado más evidencias de un defecto de la fibrinólisis y de presencia de actividad antifibrinolítica. Las Shiga-toxinas disminuyen el activador tisular del plasminógeno tipo urokinasa en cultivos de células endoteliales, y los lipopolisacáridos y el TNF-a disminuyen el t-PA, mientras aumentan el PAI-1 . Además, las Shiga toxina-1 disminuyen el PAI-1 y t-PA después de la pre-incubación con TNF-a, lo cual probablemente se debe a una disminución general en la síntesis de proteínas en cultivos de células endoteliales. En conclusión, la actividad profibrinolítica de las células endoteliales podría estar disminuida en el SHU, lo cual predispone a la trombosis microvascular.26
La existencia de casos familiares y recurrentes sugiere que puede haber un trasfondo genético de susceptibilidad a la enfermedad, de la cual, sin embargo, no se han podido identificar marcadores. 27
Cuadro clínico
La infección por E. coli 0157:H7 puede manifestarse como una infección asintomática o llegar a causar la muerte, pasando por cuadros diarreicos sanguinolentos, no sanguinolentos, PTT o SHU. Se estima que aproximadamente el 10 % de las infecciones por E. coli productor de verotoxinas, en niños menores de 10 años, evolucionan hacia el SHU. 28
Este cuadro suele verse precedido por diarrea sanguinolenta, fiebre elevada e importante dolor abdominal tipo cólico. Existen una serie de factores, cuya presencia hace más probable la evolución de una infección por E. coli hacia un SHU y que por lo tanto, son un signo de alarma:
- Fiebre elevada (temperatura rectal mayor de 39 °C).
- Importante leucocitosis (mayor de 13X109).
- Diarrea sanguinolenta.
- Edad inferior a 2 años.
- Tratamiento previo con agentes espasmolíticos.
La tríada clínica fundamental de SHU/PTT consiste en:
a) Insuficiencia renal aguda.
b) Anemia hemolítica microangiopática.
c) Trombocitopenia.
El daño del endotelio glomerular y la activación plaquetaria secundaria hacen que estas células se hinchen, se separen de la membrana basal y formen un espacio subendotelial. Todo esto provoca una disminución de la luz capilar glomerular, que puede favorecerse por la liberación de sustancias vasoactivas (citocinas) por las células endoteliales y las plaquetas, que tienen una potente acción vasoconstrictora, lo que provoca un aumento de la resistencia vascular renal y disminuye el flujo sanguíneo renal. De esta manera, se reduce la superficie de filtración y se alteran las propiedades de filtrado de la membrana con la consiguiente disminución del filtrado glomerular (FG). Además, la infiltración de polimorfonucleares puede liberar proteasas que aumentan el daño a las células endoteliales y a la membrana basal.29 También se ha descrito una alta concentración de uratos en el suero, que puede explicarse además de la IRA, por una nefropatía por uratos sobreañadida a la oligoanuria, causada por el daño glomerular, que podría tener un componente tubular atribuible a esta nefropatía.
De forma clásica se ha considerado que la insuficiencia renal, componente esencial del síndrome, es más grave en el SHU que en la PTT, pero existen numerosas excepciones. Así, se ha podido comprobar que entre el 40 y 80 % de los casos con PTT, presentan una afectación renal significativa.11
El grado de insuficiencia renal es muy variable, pero puede llegar a ser importante y requerir diálisis. Este suele ser el caso en el SHU, mientras que en el enfermo con PTT, la afección renal es habitualmente más moderada.
Suelen manifestarse también otros signos de afectación del riñón, como hematuria macroscópica, proteinuria de hasta 3g/24 h y presencia de cilindros hialinos, granulosos o hemáticos en el sedimento urinario.
Junto con la insuficiencia renal, un hallazgo frecuente es la hipertensión arterial grave y a veces maligna, probablemente a causa de la lesión microvascular y la consiguiente activación del sistema renina-angiotensina. En la actualidad se están observando casos secundarios a la infección neumocócica llamados casos atípicos, ya que no están asociados generalmente con diarreas, de los cuales se piensa que tengan una patogenia diferente y generalmente afecta a niños menores de 2 años.19
Algunos pacientes desarrollan insuficiencia cardíaca y anomalías electrocardiográficas. Pueden influir varios factores como la hipertensión, la retención hidrosalina secundaria a la insuficiencia renal y a la anemia, pero se piensa que existe una miocardiopatía específica ligada al síndrome. Clásicamente también se consideraba que la afectación neurológica era propia de la PTT y que estos rasgos diferenciaban la PTT del SHU. Sin embargo, este puede asimismo manifestarse con sintomatología neurológica. Las manifestaciones neurológicas del SHU/PTT pueden presentarse en forma de desorientación, confusión, convulsiones, signos focales o coma. La enfermedad puede cursar con fiebre, en particular en los casos de PTT.
El segundo componente esencial del SHU/PTT es la anemia hemolítica microangiopática; siempre se ha planteado que esta es secundaria a la disminución de la luz de los pequeños vasos en relación con el aumento de tamaño de las células endoteliales lesionadas y la formación de microtrombos. De esta manera, al pasar los eritrocitos por la luz disminuida de estos vasos lesionados, se dañan y fragmentan, adquiriendo formas abigarradas (esquistocitos), y son posteriormente retirados de la circulación por el sistema reticuloendotelial (SRE). Sin embargo, es probable que existan factores eritrocitarios que contribuyan a la aparición de la anemia.30
Recientemente se ha descrito que en la membrana de los eritrocitos jóvenes existen receptores que a través de otras sustancias (multímeros grandes de factor vW, trombospodina, etc.), sirven para que estos se adhieran a las células endoteliales dañadas. De esta manera, estos eritrocitos adheridos al endotelio se rompen con facilidad, fundamentalmente por la presión anormalmente alta existente en este nivel debida a la disminución de la luz de los vasos sanguíneos.31
Para su diagnóstico, es esencial observar en la extensión de sangre periférica hematíes fragmentados, esquistocitos y células en casco. A menudo se encuentran otros signos de hemólisis: reticulocitosis, descenso de la haptoglobina, hiperbilirrubinemia indirecta y aumento de las cifras de la enzima láctico deshidrogenasa (LDH).
El tercer componente de la tríada es la trombocitopenia causada por el consumo periférico. En la PTT el recuento de plaquetas se encuentra muy disminuido, con cifras habitualmente inferiores a 30 X109 /L; en cambio, en el SHU la plaquetopenia suele ser más moderada, la cifra de plaquetas puede estar próxima a la normalidad (alrededor de 100 X109 /L), o bien hallarse más comprometida. Como resultado de la intensa trombocitopenia de la PTT, los pacientes pueden presentar púrpura cutánea, hemorragias retinianas, gingivales, digestivas, metrorragias, epistaxis o hematuria. La evidencia indica que tanto en el SHU como en la PTT, los estudios de coagulación suelen ser normales, con la posible excepción de un discreto aumento de los productos de degradación del fibrinógeno (PDF). Un comentario especial merecen las formas de SHU/PTT asociadas con diversas complicaciones del embarazo, la ingestión de anovulatorios o el posparto, las cuales podrían estar causadas por la liberación de sustancias procoagulantes en la circulación, como la tromboplastina, que activarían el sistema de coagulación e inducirían la formación de microtrombosis en la pequeña circulación. Estas mujeres desarrollan el cuadro típico de SHU/PTT tras accidentes obstétricos (aborto séptico, retención de fragmentos placentarios, abrupto placentae), bien después de días, semanas o meses de un parto normal o, simplemente, tras tomar anovulatorios durante períodos que pueden oscilar entre 1 y 10 años. El pronóstico de esta forma de SHU/PTT parece ser particularmente malo; las pacientes que sobreviven no recuperan la función renal y suelen requerir tratamiento sustitutivo mediante diálisis o trasplante renal. 31
Diagnóstico
Los criterios diagnósticos tradicionales incluyen: anemia hemolítica microangiopática, trombocitopenia y la insuficiencia renal aguda. El diagnóstico es simple y fácil si esta tríada ocurre en un niño después de un episodio de diarrea, y sobre todo si es sanguinolenta.
Los síndromes incompletos también ocurren; la anemia y la trombocitopenia pueden ser ligeras o estar ausentes, y la nefropatía aguda puede no existir o ser ligera.
Se debe considerar el diagnóstico de infección por E. coli 0157:H7 en todo paciente con diarrea sanguinolenta o el SHU. El diagnóstico debe considerse también en personas con diarreas no sanguinolentas que pueden haber estado expuestos al microorganismo.
Se deben realizar coprocultivos en búsqueda de E. coli 0157:H7.
Aunque entre el 80 y 90 % de la E. coli de las heces del humano no fermentan en sorbitol rápidamente, se puede utilizar el agar de sorbitol-MacConkey como medio de cultivo. Es fundamental recoger las heces lo más rápidamente posible, debido al corto período de excreción del germen. Por este motivo, en las heces de los pacientes con SHU no suele aislarse el agente causal una vez que este ya se ha desarrollado (lo cual suele ocurrir un promedio de 7 días después de comenzar los síntomas de gastroenteritis), por lo que el diagnóstico de SHU suele confirmarse con el estudio de anticuerpos séricos frente al lipopolisacárido de E. coli 0157:H7.32
Las colonias que se muestran incoloras sorbitol negativo, pueden ser detectadas por el anti-suero disponible comercialmente para la 0:157. La E. coli sorbitol negativa que aglutina en anti-suero O:15, puede ser presumiblemente identificada como E. coli 0157:H7, y quedaría pendiente la identificación de H:7 en otro laboratorio especializado.
Las pruebas serológicas para la detección de anticuerpos contra 0:157 se realizan solo en laboratorios de investigación.
Diagnóstico diferencial
El SHU/PTT debe diferenciarse de otros procesos que cursan con plaquetopenia, como el lupus eritematoso sistémico, la púrpura trombocitopénica inmunológica y la coagulación intravascular diseminada. Otros procesos que pueden confundirse con el SHU/PTT son la eclampsia, la glomerulonefritis aguda, la necrosis cortical y diversos tipos de vasculitis. Puede ser muy difícil distinguir el SHU/PTT de la hipertensión maligna con anemia hemolítica microangiopática. La diferencia con la PTT es que esta es más frecuente en adultos, y aunque ambas pueden ser iniciadas por la E. coli 0157: H7, es raro un síndrome diarreico en la PTT. Generalmente el cuadro se acompaña de fiebre y trastornos neurológicos, los trastornos renales son raros y la mortalidad y recurrencia son mayores.
Lo mismo cabe decir de la esclerodermia, aunque en esta los cambios cutáneos suelen orientar el diagnóstico. Cuando la hipertensión maligna o la esclerodermia cursan con anemia microangiopática, no suele haber trombocitopenia. Las formas de SHU/PTT asociadas con enfermedades glomerulares facilitan el diagnóstico, puesto que en la hipertensión maligna y la esclerodermia las lesiones glomerulares son escasas.
Exámenes complementarios
- Hemograma: anemia de grado variable, que puede llegar hasta valores entre 4 y 5g/dL. Esta es secundaria a una hemólisis intravascular con prueba de Coombs casi siempre negativa. En la lámina de sangre periférica se observan fragmentocitos debido a que esta anemia es de tipo microangiopática. Hay una reticulocitosis presente.
Se observa trombocitopenia en el 90 % de los casos, la cual es secundaria a una destrucción periférica. Esta es menos frecuente en el SHU que en la PTT. La sobrevida plaquetaria y eritrocitaria está acortada.
Con frecuencia los pacientes presentan una leucocitosis con neutrofilia y el recuento leucocitario puede ser superior a 30X109/L.
- Medulograma: solo muestra respuesta compensatoria, con hiperplasia de los sistemas eritropoyético y megacariopoyético.
- Química sanguínea: como reflejo de una hemólisis, se puede detectar una hiperbilirrubinemia indirecta (rara vez excede los 2 a 3mg/dL), así como una disminución de la haptoglobina. También encontramos una elevación de la LDH, cuya persistencia indica que la hemólisis continúa.
- Pruebas de función renal: como consecuencia de la IRA, la urea y la creatinina se elevan, pueden aparecer trastornos electrolíticos como hipercalcemia, hiperfosfatemia, hiperuricemia, hiponatremia, hipocalcemia e hipocarbonatemia.
Casi siempre se detecta un síndrome de proteinuria-hematuria, con presencia a veces de un síndrome nefrótico.
- Alteraciones de la coagulación: estas alteraciones son poco llamativas; se observa disminución del fibrinógeno y presencia de los productos de degradación de la fibrina (PDF). El tiempo de protombina (TP) y el tiempo parcial de tromboplastina o Kaolín, suelen ser normales.
- Actividad de renina plasmática: está elevada, y contribuye, en parte, a la instauración de la hipertensión arterial.
- Complemento: en algunos casos se han detectado alteraciones, con concentraciones disminuidas de C3, C4 y CH50.
- Proteinograma: al comienzo de la enfermedad, la IgG puede estar disminuida, mientras que la IgA e IgM están elevadas.
- Heces fecales: crecimiento de la E. coli 0157: H7, así como la detección de la toxina en heces fecales y sangre.
- Orina: se puede observar hematuria, leucocituria, proteinuria y cilindruria.
Tratamiento
La indicación y el tratamiento específico destinados a prevenir o limitar la cascada de eventos que terminan en la trombosis intravascular e injuria tisular, difieren sustancialmente de los niños con SHU a los adultos con SHU/PTT. Los niños con SHU asociados a la Shiga-toxina de la E.coli, generalmente se recuperan espontáneamente y no requieren terapia con plasma. La corrección de las alteraciones hidroelectróliticas y el reposo intestinal, son el tratamiento de elección en los casos de SHU D+, con recuperación espontánea en algunas semanas,que se observa entre el 85 y 95 %.33 Por el contrario, existe un consenso general en los casos de SHU/PTT del adulto, que plantea que el recambio o la infusión de plasma deben ser siempre intentados para minimizar el riesgo de muerte o secuelas a largo plazo.33 La evolución de las formas secundarias, depende principalmente del pronóstico de la condición subyacente.
La terapia específica debe comenzar tan pronto como se establezca el diagnóstico, con la intención de controlar rápidamente la enfermedad y minimizar el riesgo de secuelas. El recuento de plaquetas y el nivel sérico de la LDH son los marcadores más sensibles para monitorear la respuesta a la terapia con plasma. El tratamiento debe continuarse hasta la completa remisión de la enfermedad. No obstante, no existen parámetros clínicos que puedan predecir de modo preciso la duración del tratamiento.
La mortalidad de esta entidad se ha reducido significativamente en los últimos 40 años de un 40 al 50 % a un actual 3 al 5 %, probablemente como resultado de un mejor manejo de las medidas de soporte, control de la anemia, insuficiencia renal, hipertensión arterial y disbalance del agua y electrolitos. No obstante, no existe ninguna terapia que se haya mostrado eficaz para prevenir o limitar el proceso microangiopático y consecuentemente, afectar el curso de la enfermedad. Los agentes antidiarreicos pueden incrementar el riesgo de megacolon tóxico. 34 El uso de antibióticos para tratar la infección por E. coli 0157:H7, ha mostrado incrementar el riesgo de SHU 17 veces. 35 La injuria a la membrana bacteriana inducida por el antibiótico podría favorecer la liberación de grandes cantidades de toxina preformada, sumado a que la terapia antibiótica podría dar a la E. coli 0157:H7 una ventaja selectiva si estos organismos no son eliminados tan rápidamente del intestino como lo es la flora intestinal normal.
Además, ciertos antibióticos, en particular las quinolonas, trimetopin y flurazolidone, son potentes inductores de expresión del gen de la Shiga-toxina 2 y pueden incrementar el nivel de toxina en el intestino. 34 Estas consideraciones no necesariamente se aplican a muchos casos de diarrea sanguinolenta, en particular en Sudamérica e India, que son precipitadas por cepas de E. coli diversas a la 0157:H7 o por otras bacterias, tales como la Shigella dysenteriae tipo 1, en las que el uso de un tratamiento antibiótico empírico acorta la duración de la diarrea, disminuye el índice de complicaciones y reduce el riesgo de transmisión de la enfermedad. De tal manera, en los países en desarrollo, donde la Shigella es el agente causal más frecuente de colitis hemorrágica, la terapia antibiótica empírica debe iniciarse tempranamente, aún antes que el patógeno involucrado sea identificado. 36
Se encuentran bajo evaluación nuevos agentes destinados a prevenir la exposición de los órganos a la Shiga-toxina. El más promisorio es una resina, Synsorb-Pk (Chromosorb) compuesta por carbohidratos sintéticos unidos a una sílice coloidal que se une con la Shiga-toxina; otro es la E. coli modificada por recombinación genética, que muestra sobre su superficie un receptor que se asemeja al receptor para la Shiga-toxina, que absorbe y neutraliza la Shiga-toxina con alta eficiencia; por último, el STARFISH, que es un carbohidrato oligovalente soluble en agua que puede simultáneamente ligar las 5 subunidades B de 2 moléculas de toxina. 37
Estas terapéuticas ofrecen nuevas y potentes armas contra la Shiga-toxina de la E. coli inductora de colitis hemorrágica y SHU. La administración oral de Synsorb podría limpiar eficientemente de toxina el intestino, en tanto que la administración endovenosa de STARFISH, podría ayudar a prevenir que la toxina presente en circulación afecte los riñones y la microcirculación. El análisis preliminar de un estudio canadiense que se encuentra en curso, encontró que el tratamiento precoz (dentro de los 2 primeros días de iniciada la diarrea con Synsorb-Pk), disminuye el riesgo del SHU del 17 al 7%.37
La transfusión de plaquetas está contraindicada, pues produce un empeoramiento del estado del paciente debido al daño orgánico que puede producir. 38
La infusión de inmunoglobulinas inhibe algunos factores agregantes encontrados en el plasma de algunos pacientes y tiene un posible rol en la neutralización de las verotoxinas. 39
Los antiagregantes plaquetarios (ácido acetilsalicílico, dipiridamol, sulfinpirazona), tienen un efecto beneficioso, sobre todo en la PTT del adulto, pero su eficacia en el SHU es muy inferior. Se dice que no son beneficiosos cuando se utilizan solos, deben usarse junto con la plasmaféreis. La heparina y los fibrinolíticos no se han demostrado eficaces y pueden ser peligrosos, ya que aumentan el riesgo de sangramiento, por lo que no se usan en la actualidad.
En la PTT, varias combinaciones de antiagregantes plaquetarios junto con glucocorticoides y esplenectomía en casos resistentes, permiten alcanzar supervivencias del 50 al 60 %. En casos refractarios se ha utilizado la vincristina. 40
Los resultados son igualmente buenos con la perfusión de plasma, cuyo objetivo es proporcionar el factor estimulador de la PGI2. La plasmaféresis presenta, en teoría, ventajas sobre la perfusión de plasma en el tratamiento del SHU/PTT, ya que además de administrar plasma, se eliminan sustancias circulantes que podrían tener un papel patogénico importante, así como evita la sobrecarga de volumen. Sin embargo, no hay estudios controlados sobre su eficacia. Es posible que la administración de plasma sea beneficiosa sólo en adultos con SHU/PTT y quizás en niños con formas hereditarias, que cursan con bajos niveles de PGI2.41
El tratamiento con vitamina E se ha utilizado para corregir el reducido poder antioxidante detectado en el SHU y posiblemente involucrado en el daño peroxidativo de los hematíes. La vitamina E administrada en una dosis de 1 000 mg/m2/día durante al menos una semana, se ha asociado con un buen curso clínico en niños con SHU.42
La perfusión de PGI2 ha proporcionado resultados contradictorios.
Los enfermos que desarrollan insuficiencia renal requieren diálisis o trasplante. 43
En la tabla 2 se muestran las pautas terapéuticas utilizadas en el SHU/PTT y su forma de administración.
Tabla 2. Tratamiento más comunmente usado en el síndrome hemolítico urémico/púrpura trombocitopénica trombótica (SHU/PTT),dosis y modalidades de administración
| Tratamiento | Administración | Indicación/Comentarios |
| Tratamiento de la IRA | SHU | |
| Diálisis peritoneal | Continua, 24 horas | Bien tolerada puede eliminar el activador tisular del plasminógeno 1 |
| Adsorción verotoxina | Oral 0,5g/Kg x 7 días | SHU |
| Chromosorb | Limita absorción de verotoxina y lesiones microangiopáticas (bajo investigación clínica) | |
| Agentes anti plaquetarios | PTT | |
| Aspirina | Oral 325-1 300 mg/día | Eficacia no probada |
| Dipiridamol | Oral 400-600 mg/día | Eficacia no probada |
| Dextran 70 | EV 500 mL 2 veces al día | Eficacia no probada |
| Prostaciclina | EV 4-20 µg/kg/min | Beneficio en algunos casos ocasionales |
| Agentes antitrombóticos | SHU | |
| Heparina | EV 5 000 | Se han visto beneficios en |
| Estreptoquinasa | EV 2 500 U EV 100 000 U/hora |
Eficacia no probada. Produce |
| Esteroides / Vincristina | PTT | |
| Prednisona | Oral 60-200 mg/día | PTTPosible efecto en formas ligeras |
| Vincristina | EV 1mg/4 días | Efecto posible en recaídas |
| Plasma fresco congelado | SHU atípico y del adulto y PTT | |
| Infusión | 30-40 mL/kg el día 1, después 10-20 mL/kg/día | Efectivo en casos atípicos y en trastornos neurológicos |
| Plasmaféresis | 1-2 vólumenes plasmáticos | Igual que en la infusión, sin riesgo de sobrecarga volumen |
| Otros tratamientos | SHU | |
| Vitamina E | Oral 1 000 mg/m2/día | Efectivo en SHU típico |
| Gammaglobulina | EV 400 mg/kg/día | SHU o PTT |
| Terapia de rescate | ||
| Esplenectomía | Efectivo en PTT en recaída ocasionalmente | |
| Nefrectomía bilateral | Efectivo en SHU resistente al plasma |
Manejo del paciente con SHU
- Control estricto del equilibrio hidromineral con restricción de los líquidos a 400 mL/m2 y manejo de la hiponatremia.
- Manejo de la anemia, solo se tratará si existen signos o síntomas de hipovolemia o Hb inferior a 70 g/L.
- No uso de antibióticos de forma rutinaria. Estudios actuales revelan la liberación de toxinas (Shiga-like) después de la administración de antibióticos. 44
- Si el ritmo diurético es inferior a 1mL/kg/h, utilizar infusión de furosemida. 45
- Dopamina a dosis renales, con el objetivo de mejorar la perfusión a este nivel.
- Control estricto de la tensión arterial.
- Diálisis peritoneal, con las indicaciones siguientes:
- Oliguria que no responde a dosis alta de furosemida.
- Sobrecarga hídrica.
- Acidosis que no responde a la administración de bicarbonato.
- Hipertensión arterial refractaria a las medidas farmacológicas.
En casos que sea difícil o esté contraindicada la diálisis peritoneal, se recomienda el uso de hemodiálisis. En casos extremos se ha llegado al trasplante renal, cuando el paciente evoluciona a insuficiencia renal crónica.
Pronóstico
- Aproximadamente el 85 % de los niños con SHU se recuperan completamente con terapia de soporte.
- Aproximadamente del 15 al 20 % de los niños pueden presentar hipertensión de 3 a 5 años, después de la aparición de la enfermedad.
- En adultos con SHU la mortalidad es baja, sin embargo, la función renal puede tener un pronóstico pobre en pacientes no tratados.
- Se estima que cerca del 80 % de los adultos con SHU, pudieran requerir diálisis o trasplante renal a largo plazo.
Criterios de mal pronóstico
Se han expuesto una serie de factores de mal pronóstico en el SHU, los cuales se muestran a continuación:
- Insuficiencia renal prolongada.
- Presencia creciente de daño renal. Compromiso glomerular extenso (>80 %).
- Edad superior a 5 años.
- Compromiso de arterias de mediano calibre.
- Consumo persistente de factores de la coagulación.
- Hipertensión arterial severa (especialmente aparición tardía).
- Compromiso importante del SNC.
- Niños mayores que desarrollan la enfermedad en los meses de invierno.
- Pacientes con cáncer que reciben quimioterapia.
Summary
It was made a review of the general aspects of the uremic haemolytic syndrome (UHS): epidemiology, pathogeny, clinical manifestations and treatment. UHS results from the action of numerous ethiological and pathogenic factors It is mainly caused by a verotoxin produced by different Escherichia coli strains. In this entity, it is produced an endothelial damage that generates a series of phenomena, such as adhesion, platelet aggregation and deposits of fibrin, leading to the formation of thrombi in the microcirculation: thrombotic microangiopathy. UHS is characterized by a combination of acute renal failure, thrombocytopenia and microangiopathic haemolytic anemia. Diarrheas and upper respiratory tract infections are the most common precipitating factors. Plasmaphaeresis and dialysis are nowadays the election treatment.
Subject headings: HEMOLYTIC-UREMIC SYNDROME/ epidemiology; HEMOLYTIC-UREMIC SYNDROME/ etiology; HEMOLYTIC-UREMIC SYNDROME/ drugtherapy; ESCHERICHIA COLI; KIDNEY FAILURE, ACUTE; TROMBOCYTOPENIA; ANEMIA, HEMOLYTIC; CHILD.
Referencias bibliográficas
- Ruggenenti P, Remuzzi G. Thrombotic thrombocytopenic purpura and related disorders. Hematol Oncol Clin North Am 1990;4:219.
- Gasser C, Gautier E, Steck A, Siebenmann RE, Dechslni R. Hamolytisch-uramische syndromes bilaterale Nierenrindennekrosen bei akuten erworbenchen hamolytischen Anamien. Schweiz Med Wschr 1995;85:905-9.
- Moschocowitz E. Acute febrile pleiochromic anemia with hyaline thrombosis of terminal arterioles and capillaries: an undescribed disease. Arch Intern Med 1925 ;36 :89-93.
- Neil MA. Pathogenesis of Escherichia coli 0157H7 infection. Curr Op Infect Dis 1994;7:295-303.
- Tumlin JA, Sands FM, Someren A. Special feature: Hemolytic uremic syndrome following "Crack Cocaine" inhalation. Am J Med Sci 1990; 299: 366-70.
- López EL, Díaz M, Gainstein S, Devoto S, Mendilaharzu F. Hemolytic uremic syndrome and diarrhoea in Argentine children. The role of Shiga- Like toxins. I. Infect Dis 1989;160:469-75.
- CDC. Foodborne diseases active surveillance network (FoodNet): population survey atlas of exposures: 1996--1997. Atlanta, Georgia: US Department of Health and Human Services, CDC; 1997.
- Remuzzi G. Thrombotic thrombocytopenic purpura and allied disorders .En: Verstraete M, Vermylen J, Lunen HR, Arnout J, eds. Thrombosis and Haemostasis. Leuven: Leuven University Press;1987: 673-708.
- Pearson JD. Endothelial cell function and thrombosis. Baillieres Clin Haematol 1994;7:441-52.
- Caletti MG, Gallo G, Gianantonio CA. Development of focal segmental sclerosis and hyalinosis in hemolytic uremic syndrome. Pediatr Nephrol 1996;10:687-92.
- Corrigan Jr. JJ, Boineau FG. Hemolytic-uremic syndrome. Pediatr Rev 2001; 22(11):365-9.
- Karmali MA, Petric M, Lim C, Fleming PC, Steele BT. Escherichia Coli-cytotoxin, hemolytic uremic syndrome and haemorragic colitis. Lancet 1983;2:199.
- Beatle TL. Recent development in the pathogenesis of hemolytic uremic syndrome. Renal Fail 1990;12:3-7.
- Benigni A, Remuzzi G. The role of eicosanoids in pathogenesis of hemolytic uremic syndrome . Prostaglandins Leukot Essen. Fatty Acids 1994;51:75-9.
- Caprioli J, Bettinaglio P, Zipfel PF. The molecular basis of familial hemolytic-uremic syndrome: Mutation analysis of factor H gene reveals a hot spot in short consensus repeat 20. J Amer Soc Nephrol 2001; 12: 297-307.
- Zipfel PF. Haemolytic uremic syndrome: How do factor H mutants mediates endothelial damage? Trends Immunol 2001;22:345-8.
- Capriolo J, Bettinaglio P, Zipfel PF. The molecular basis of familiar hemolytic uremic syndrome :mutation analysis of factor H gene reveals a hot spot in short consensus repeat 20 . J Am Soc Nephrol 2001; 12:297-307.
- Shiomi M, Togawa M. Sporadic cases of hemolytic uremic syndrome and hemorragic colitis with serum IgM antibodies to lipopolysaccharides of enterohemorrhagic Escherichia coli O157H7. Nippon Rinsho 1997;55(3):686-92.
- Mc Ligeyo SO. Haemolytic uraemic syndrome: a review. East Afr Med J 1999;76(3):148-53.
- Forsthy KD, Simpson AC. Neutrophil-madiated endothelial injury in haemolytic uremic syndrome . Lancet 1989;2:411-4.
- Milford DV, Staten J, MacGreggor I. Prognostic makers in diarrhoea-associated Haemolytic uremic syndrome: initial neutrophil count , human neutrophil elastase and von Willebrand factor antigen . Nephrol Dial Transp 1991;6:232-7.
- Fong JSC, Kaplan BS. Impairment of platelet aggregation in the Hemolytic uremic syndrome: evidence of platelet "exhaution" . Blood 1982;60:564-70.
- Van Hinsberg VW. Regulation of the synthesis and secretion of plasminogen activators and plasminogen activator inhibitor by endothelial cells. Haemostasis 1998;18:307-27.
- Furlan M, Lammle B. A. Etiology and pathogenesis of thrombotic thrombocytopenic purpura and haemolytic uraemic syndrome: the role of von Willebrand factor-cleaving proteasa. Best Pract Clin Haematol 2001;14(2):437-54.
- Cid AR, De la Rubia J, Aznar JA. Importancia de la proteasa escindidora del factor von Willebrand en la fisiopatología, diagnóstico y tratamiento de la púrpura trombótica trombocitopénica. Rev Iberoamer Tromb Hemostasia 2001;14(2):63-7.
- Van de Kar NC, Van Hinsbergh VW, Brommer EJ, Monnens LA. The fibrinolytic system in the hemolytic uremic syndrome: in vivo and in vitro studies. Pediatr Res 1994;257-64.
- Niaudet P, Gagnadoux MF, Broyer M. Hemolytic-uremic syndrome: hereditary forms and forms associated with hereditary diseases. Adv Nephrol Necker Hosp 2000;30: 261-80.
- Neild GH. Hemolytic uremic syndrome in practice. Lancet 1994; 343:398-401.
- Remuzzi G. The hemolytic-uremic syndrome. Perspective in clinical nephrology. Kidney Int 2000;47:2-19.
- Ruggenenti P, Remuzzi G. The pathophysiology and management of thrombotic thrombocytopenic purpura. Eur J Haematol 1996;56:191-207.
- Malvinder S Parmar. Hemolytic-uremic syndrome. Med J 2001;2(11).
- Bitzan M, Moebious E, Ludwing K, Muller-Wiefeld DE. High incidence of serum antibodies to Escherichia Coli O 157 lipopolisacharide in children with Hemolytic Uremic Syndrome. J Pediatr 1991;119:380-4.
- George JN. How I treat patients with thrombotic thrombocytopenic purpura- hemolytic uremic syndrome. Blood 2000;96:1223-9.
- Zhang X, McDaniel AD, Wolf LE. Quinolone antibiotics induce Shiga toxin-encoding bacteriophages, toxin production, and death in mice. J Infect Dis 2000;181:664-70.
- Wong CS, Jelacic S, Habeeb RL. The risk of hemolytic uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N Engl J Med 2000;342:1930-6.
- O'Ryan M; Prado V. Risk of the hemolytic uremic syndrome after antibiotic treatment of Escherichia coli 0157:H7 infections. N Engl J Med 2000;343:1271.
- Paton AW, Morona R, Paton JC. A new biological agent for treatment pf Shiga toxigenic Escherichia coli infections and dysentery in humans. Nat Med 2000;6:265-70.
- Gordon LI, Kwaan HC, Rossi EC. Deleterious effects of platelet transfusions and recovery thrombocytosis in patients with thrombotic microangiopathy. Semin Hematol 1987; 24(3): 194-201.
- Loirat C, et al. Treatment of childhood hemolytic-uremic syndrome with immunoglobulins (abstract). Ped Nephrol 1991; 5: C39.
- Van Gool S, Brock P, Van Laer P. Successful treatment of recurrent thrombotic thrombocytopenic purpura with plasmapheresis and vincristine. Eur J Pediatr 1994; 153: 517-9.
- EI-Reshaid K, Kapoor MM, Nampoory MR, EI-Reshaid W Johny KV. Pediatric dialysis and renal transplantation in Kuwait over the past 11 years. Ped Nephrol 1999;13(3):259-64.
- Powell HR , McCredie DA, Taylor CM, Burke JR, Walker RG. Vitamin E treatment of haemolytic uraemic syndrome. Arch Dis Child 1984;59:401-4.
- Ruggenenti P, Remuzzi G. Treatment of adult hemolytic-uremic syndrome. Adv Nephrol Necker Hosp 2000; 30: 83-94.
- Zimmerhack LB. E. coli, antibiotics, and the hemolytic uremic syndrome. N Engl J Med 2000;342:1990-1.
- Ben Ezer D, Veiga PA, James JA. Intravenous furosemide infusion decreases the need for dialysis with hemolytic uremic syndrome(HUS). Ped Res 1994; 35:361.
Recibido: 10 de agosto del 2003. Aprobado: 17 de agosto del 2003.
Dr. Juan Carlos Jaime Fagundo. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800, Ciudad de La Habana, Cuba. Tel (537) 578268, Fax (537) 442334. e-mail: ihidir@hemato.sld.cu
1 Instituto de Hematología e Inmunología. Ciudad de La Habana, Cuba.
2 Hospital Docente "José Ramón Martínez'' , Guanajay. La Habana, Cuba.
3 Hospital General Universitario "Comandante Pinares", San Cristóbal. Pinar del Río, Cuba.
4 Hospital Pediátrico Docente "William Soler''. Ciudad de La Habana,Cuba.

