










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter v.21 n.3 Ciudad de la Habana sep.-dic. 2005
Instituto de Hematología e Inmunología
Nuevos conocimientos sobre el metabolismo del hierro
MsC. Mariela Forrellat Barrios, Dra. Norma Fernández Delgado y Dr. Porfirio Hernández Ramírez
Resumen
El hierro es un mineral de elevada importancia para el organismo y su regulación requiere de una red molecular compleja. Hasta hace unos años solo se conocían 3 proteínas que intervenían en el metabolismo del hierro, pero en la última década, se han descubierto de forma secuencial, y a partir del estudio de algunas enfermedades genéticas como la hemocromatosis hereditaria, nuevas proteínas que participan en la homeostasia del hierro y que están implicadas en su transporte, absorción, reciclaje y balance en el organismo. La identificación y aislamiento de estas proteínas lleva inevitablemente a la modificación de los modelos clásicos de regulación de la homeostasia de este importante mineral. En este trabajo se realizó una revisión de los elementos esenciales conocidos hasta la actualidad de cada una de estas nuevas proteínas y la interacción entre ellas.
Palabras clave: hierro, hepcidina, HFE, hemojuvelina .
El hierro es un elemento muy abundante en la naturaleza y esencial para la vida de prácticamente todas las formas biológicas. Contradictoriamente, las mismas propiedades que hacen a este metal de transición imprescindible para procesos biológicos vitales, como el transporte de oxígeno y de electrones, lo hacen tóxico, pues es capaz de generar radicales libres que provocan daño oxidativo de importantes componentes celulares. No es raro entonces, que durante el proceso evolutivo se hayan desarrollado mecanismos que permiten mantener un estricto control de los niveles de este mineral.
Para mantener la homeostasia del hierro, los organismos deben ser sensibles a los cambios en sus niveles y responder a ellos alterando los procesos de absorción y almacenamiento del mineral. En los humanos, el control se ejerce fundamentalmente sobre la cantidad de hierro que se absorbe, más que sobre su excreción. La respuesta inadecuada o la pérdida de respuesta ante las variaciones de los niveles de hierro conducen a la anemia o a su sobrecarga.1
El control del metabolismo del hierro en los mamíferos requiere de una red molecular compleja y estrictamente regulada. En los últimos años se han producido importantes avances en el campo de este metabolismo, que han modificado la visión clásica del mismo, como consecuencia del descubrimiento de un número importante de nuevas moléculas proteicas. Entre estas proteínas se encuentran transportadores de hierro ferroso como el transportador de metales divalentes 1(DMT1) y la ferroportina; enzimas con actividad ferrooxidasa como la hefastina y la ceruloplasmina, o ferrirreductasa como el citocromo b duodenal y proteínas reguladoras como HFE (contracción del término en inglés relacionado con HLA-H, que es la región del sistema HLA cercano al gen y FE como símbolo del hierro), hepcidina y hemojuvelina (HJV), así como un segundo receptor de transferrina ( RTf2 ). 2 El descubrimiento de estas proteínas, así como la vinculación de las alteraciones de su síntesis con estados patológicos del metabolismo férrico, han conducido a una mejor comprensión de este metabolismo y han modificado los modelos previos de regulación de la homeostasia del mineral, aspectos estos que serán expuestos en el presente trabajo.
Nuevas proteínas en el metabolismo del hierro
La homeostasia del hierro debe ser controlada estrictamente. Hasta hace algo más de una década solo se conocían 3 proteínas que intervenían en el metabolismo del hierro: la ferritina (principal proteína de reserva), la transferrina (principal transportador) y el receptor de transferrina ( RTf ), indispensable para la internalización del mineral. Con el descubrimiento de las proteínas reguladoras de hierro (conocidas por sus siglas en inglés, IRP) capaces de unirse a los elementos de respuesta al hierro o IRE (del inglés iron responsive elements) de los ARN mensajeros (ARNm) de las proteínas implicadas en el metabolismo del mineral, se amplió el horizonte para comprender los complejos mecanismos del mantenimiento de la homeostasia del hierro.3
Los IRE son estructuras lazo-tallo localizadas en las regiones 5' o 3' no traducidas de los ARNm (5' o 3' UTR) que codifican las proteínas que intervienen en el metabolismo del hierro. Las IRP trabajan en conjunto con estos elementos para monitorear y responder a los cambios en la cantidad de hierro quelable en el ambiente intracelular conocido como compartimiento pool de hierro lábil. A través de la interacción de las IRPs con los IREs, la incorporación de hierro vía transferrina aumenta por estabilización del ARNm del RTf , mientras el almacenamiento como ferritina disminuye por bloqueo de la traducción del ARNm de esta proteína. Estos eventos resultan en un aumento del pool de hierro lábil. Inversamente, la incorporación de transferrina disminuye y el nivel de ferritina aumenta cuando la concentración intracelular de hierro es elevada (fig.1) 4,5
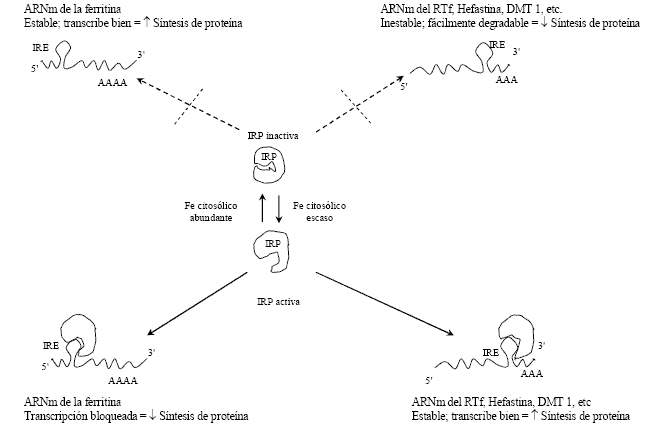
Fig. 1. Representación esquemática de la regulación del metabolismo del hierro.
IRE: elemento de respuesta al hierro; IRP: proteína reguladora de hierro; RTf : receptor de transferrina : DMT 1: Transportador de metales divalentes 1.
Recientemente, el estudio de algunas enfermedades genéticas como la hemocromatosis hereditaria y la aceruloplasminemia, han aportado nuevos elementos acerca de la función y regulación de genes implicados en el metabolismo del hierro. Así se han identificado nuevos genes y sus correspondientes proteínas que están involucrados en el transporte, absorción, reciclaje y balance del hierro en el organismo (tabla). 6
Tabla . Nuevas proteínas implicadas en el metabolismo del hierro.
| Gen/proteína | Localización del gen | Principal sitio de expresión | Función en el metabolismo del hierro
|
| HFE/HFE | 6p21.3 | Enterocitos y macrófagos | Modula la homeostasia del hierro corporal Media la incorporación celular de hierro unido a la transferrina |
| SLC11A2/ DMT 1 | 12q13 | Enterocitos | Transportador de hierro y otros metales divalentes |
| TFR2/RTf 2 | 7q22 | Hígado y células mononucleares | Sensor de la saturación de transferrina |
| Heph / Hefastina | Xq11-q12 | Intestino | Eflujo de hierro del enterocito |
| SLC40A1/ Ferroportina | 2q32 | Placenta, intestino, hígado, bazo, músculo | Exportador de hierro que participa en adquisición de hierro del medio y reciclaje de las reservas corporales |
| HAMP/ Hepcidina | 19q13 | Hígado | Hormona reguladora del hierro |
| HJV/ Hemojuvelina | 1q21 | Hígado,corazón, músculo esquelético | Modula expresión de la hepcidina |
HFE
Identificada en 1996 como producto del gen de la hemocromatosis hereditaria,7 es considerada una proteína atípica del sistema mayor de histocompatibilidad. No une péptidos ni desempeña ningún papel conocido en el sistema inmune, aunque su estructura tiene gran homología con las proteínas clase I del sistema inmune, incluida la existencia en su estructura de la hendidura o sitio para la presentación de antígenos. Tampoco une ni transporta hierro. Sin embargo, influye en su absorción, pues se asocia de forma pH dependiente con el RTf , disminuyendo la afinidad de este por la transferrina cargada, con la que compite por su unión al receptor.6,8,9
Debido a su vinculación con la vía de incorporación de hierro mediada por la transferrina y su localización en los endosomas y la cara basolateral de los precursores enterocíticos, se plantea que HFE puede ser un sensor de las reservas corporales de hierro, e incluso se ha sugerido que la relación HFE: RTf es crítica para el mantenimiento de la homeostasia del hierro.1,9
Se ha comunicado que es el gen más comúnmente mutado en la hemocromatosis hereditaria clásica. La mayoría de los pacientes con hemocromatosis tipo I son homocigotos para un alelo único que contiene la sustitución de cisteína por tirosina en el codon 282 (C282Y) de esta proteína.6,7 Esta mutación impide la formación de un puente disulfuro intramolecular crítico para la expresión de la proteína y para la interacción de esta con el RTf , lo que implica la pérdida parcial de la función de la proteína. Además, se han descrito otras mutaciones y polimorfismos del gen como son: H63D, S65C, I105T y G93R.6
En fecha reciente, se ha sugerido que HFE normalmente facilita más que obstaculiza la incorporación celular de hierro unido con la transferrina mediada por el RTf , y parece que HFE puede también unir otras proteínas o ejercer efecto directo sobre el transporte endosomal del mineral.10-13
DMT1
Esta proteína era conocida anteriormente por las siglas en inglés Nramp2 (natural resistance associated macrophage protein) o DCT1( divalent cation transporter ); es el primer transportador de hierro caracterizado al nivel molecular en mamíferos. 14 Esta transfiere el hierro a través de la membrana apical de la célula absortiva y hacia su interior a través de un proceso acoplado a protones,15,16 por lo que se plantea que actúa en 2 puntos diferentes: como transportador responsable de la absorción de hierro en el intestino y en la movilización del mineral a partir de los endosomas durante el ciclo de la transferrina, donde transporta el hierro liberado hacia el citoplasma de los precursores eritroides.14
Tiene la singularidad de no ser específico para el hierro, sino que además transporta desde la luz intestinal al interior celular otros metales pesados como manganeso, cobalto, cobre, zinc, cadmio y plomo. Sin embargo, no transporta calcio ni magnesio.16,17
La expresión de esta proteína es regulada por las reservas corporales de hierro, pero también responde al nivel de hierro dietético, y puede ser controlada por mecanismos pos-traduccionales , ya que contiene un IRE en su región 3'UTR, lo que es indicativo de que puede degradarse en el contexto de un pool de hierro libre elevado, como ocurre con el mensajero del RTf 1.
RTf2
Este receptor fue clonado en 1999 por 2 grupos independientes; es una proteína transmembrana de tipo II estructurada en un dominio citoplasmático N- terminal, un pequeño dominio transmembrana y un ectodominio C- terminal grande. Tiene el 66 % de homología con el RTf1 y el 45 % de identidad de aminoácidos con el ectodominio de este receptor.18,19
Aunque ambos receptores son capaces de transportar hierro unido a la transferrina al interior celular, sus propiedades difieren; así por ejemplo: el RTf tiene una menor afinidad por la holotransferrina que el RTf 1, con el que forma un heterodímero; además el RTf 2 no se une al HFE. El patrón de expresión del RTf 2 es elevado en el hígado y en las células mononucleares de sangre periférica, a diferencia del RTf 1, cuya expresión hepática es mucho menor.20 Resulta importante señalar la expresión del RTf 2 en líneas celulares K562 y en blastos leucémicos, especialmente de tipo M6, pues este receptor no se expresa en ninguna etapa de diferenciación en células eritroides.21
Ambos receptores difieren también en su respuesta a los cambios en el nivel de hierro celular, ya que como el ARNm del RTf 2 no contiene IRE en su región 3'UTR, sus niveles de ARNm y proteínas varían poco con los cambios en los niveles de hierro.20 Sin embargo, se plantea que puede mediar la incorporación celular de hierro en proporción directa a la saturación de transferrina plasmática, lo que implica que se comporta como un sensor de la saturación de transferrina . Además, a partir de datos experimentales, se ha planteado que contribuye a modular la producción de hepcidina,20,22 e incluso se ha sugerido que actúa por encima de la HFE y la HJV en la cascada de señales de inducción de la hepcidina.22
La función exacta del RTf 2 en el metabolismo del hierro es aún desconocida. Sin embargo, es evidente que esta es importante para el mantenimiento de la homeostasia del hierro, pues mutaciones casi todas privadas (la más conocida es Y250X) en el gen de este receptor, son la causa de una variante de hemocromatosis humana no vinculada al HFE, denominada hemocromatosis hereditaria tipo 3 (OMIM 604250, del inglés o nline M endelian i nheritance in m an).23,24
Hefastina
Se descubrió en 1999; constituye un importante punto de unión del metabolismo de 2 importantes micronutrientes: el cobre y el hierro.25 Su clonaje enfatizó la importancia del cobre en la transferencia de hierro del enterocito al plasma.1
Es una proteína rica en cobre, similar a la ceruloplasmina plasmática, con la que tiene una significativa homología estructural y probablemente funcional. Se plantea que actúa como una ferrooxidasa necesaria para el egreso de hierro del enterocito a la circulación.1,6,17,25 Su expresión es elevada en el intestino, específicamente en las vellosidades intestinales, no así en las criptas celulares, lo que confirma su papel crucial en el eflujo de hierro del enterocito al plasma. En su porción C- terminal tiene un dominio de anclaje a membrana que puede orientar la actividad ferrooxidasa sobre la superficie celular o en el interior de las vesículas, para actuar conjuntamente con un exportador de hierro.1,25
La deleción del aminoácido 194 de esta proteína es responsable del fenotipo sla (sex linked anemia) obtenido en ratones, y se plantea que previene la oxidación del hierro y el consiguiente transporte del mineral a través de la membrana basolateral del enterocito.25 Sin embargo, aún no se conoce si la actividad oxidasa de la hefastina determina la selectividad de un transportador basolateral específico o de otras proteínas transportadoras de hierro, o si mantiene un gradiente de concentración de hierro ferroso a través de la membrana basolateral.1
No obstante, se ha planteado que la hefastina forma parte, junto con el DMT1, de una vía de incorporación de hierro regulada por el HFE, y que las mutaciones en esta proteína podrían disminuir o exacerbar el fenotipo de hemocromatosis hereditaria.26
Ferroportina
Esta proteína se aisló y caracterizó en el 2000; es también conocida como Ireg1 ( iron-regulated transporter 1 ) y MTP1 (metal transporter protein). Es la primera proteína transportadora de hierro para el egreso transmembrana del mineral identificada en vertebrados. Se expresa en tejidos involucrados en la homeostasia del hierro, incluido el sistema retículo endotelial (SRE) maduro y en desarrollo, el hígado, el duodeno (especialmente en las células absortivas maduras de las vellosidades), en el útero de embarazadas y en los músculos y las células del sistema nervioso central de embriones.27-29
Este transportador es una proteína transmembrana multimérica regulada por hierro, que se localiza en la membrana basolateral de las células del epitelio duodenal y en el compartimiento citoplasmático de células del SRE, donde tiene una distribución predominantemente basolateral. No obstante, puede encontrarse en el citoplasma basal y apical de estas células. Está relacionada con la familia de los transportadores divalentes del DMT1 y como transportador de membrana de hierro ferroso requiere una actividad ferrooxidasa, para lo que se acopla a la hefastina.27
La existencia de un IRE en la región 5'UTR del ARNm de la ferroportina indica una regulación dependiente de hierro semejante a la de la ferritina. Asimismo, se ha observado que la expresión de esta proteína en hígado y duodeno es regulada por el hierro de forma recíproca y que la sobreexpresión en cultivos celulares conduce a la depleción intracelular del mineral.1,27
Se plantea que tiene una función clave en 2 aspectos diferentes de la homeostasia del hierro: la absorción del mineral por los enterocitos duodenales y la liberación de las reservas corporales por células retículoendoteliales, por lo que se postula como el principal y único exportador de hierro que funciona en estos 2 puntos claves del metabolismo férrico.1,27 Recientemente se ha planteado que la ferroportina es esencial para el reciclaje del hierro hemo por los macrófagos.30
La ferroportina además es la tercera proteína que constituye otro sitio de defecto en pacientes con hemocromatosis hereditaria,31 e incluso se describe la hemocromatosis hereditaria 4 o la enfermedad de ferroportina como entidad autosómica dominante (OMIM 606069).32 Las mutaciones más frecuentemente descritas son A77D, N144D, N144T, G323V, G490D, D157G, Q182H, entre otras.33
Hepcidina
Se aisló y purificó en el 2000 por 2 grupos independientes a partir de fluídos biológicos en los que investigaban propiedades antimicrobianas . Es una pequeña hormona peptídica de 20-25 aminoácidos perteneciente a la familia de las defensinas, péptidos antimicrobianos ricos en cisteínas . Además es un regulador central del metabolismo del hierro y se ha comprobado su implicación en desórdenes comunes de este metabolismo, como la hemocromatosis hereditaria y la anemia de los procesos crónicos.34
Esta proteína es producida exclusivamente en el hígado y secretada a la circulación en respuesta a la inflamación o al aumento de las reservas de hierro.35,36 Se ha observado que su excreción correlaciona bien con los niveles de ferritina sérica, usualmente aumentados en la sobrecarga de hierro y en la inflamación.37 Además se ha encontrado correlación inversa entre los niveles de transcripción de hepcidina y la saturación de transferrina férrica y correlación significativa con el RTF 2, independientemente del estado de hierro.38 Por otra parte, la deficiencia o ausencia de este péptido conduce a la sobrecarga de hierro y su sobreexpresión a la anemia.2
La expresión del ARNm se correlaciona con la disponibilidad de hierro para la eritropoyesis más que con las reservas del mineral, de ahí que se plantee que la hepcidina es regulada primariamente por la disponibilidad de hierro para el eritrón.39 A diferencia de otras proteínas implicadas en el metabolismo del hierro, no contiene IRE reconocible en su ARNm.40
Dentro de la homeostasia del hierro, se plantea que la hepcidina es un regulador negativo de la absorción intestinal del mineral y de la liberación del hierro por los macrófagos. No es raro entonces que sus principales dianas celulares sean los enterocitos de las vellosidades intestinales, los macrófagos del SRE y los hepatocitos.40 En relación con su papel como señal reguladora de la absorción, se conoce que sus niveles transcripcionales son afectados por los mismos cuatro factores que influyen sobre la absorción de hierro de la dieta: el nivel de eritropoyesis , las reservas de hierro, la hipoxia y la inflamación. Recientemente, se ha planteado que la hepcidina disminuye la actividad funcional de la ferroportina , con lo que controla la exportación del hierro celular. La hepcidina se une con la ferroportina e induce su internalización y degradación, lo que trae como resultado la retención celular del hierro como consecuencia de la disminución de la exportación del mineral.41
Teniendo en cuenta todas las características de esta pequeña molécula, se ha sugerido que es un excelente candidato como molécula señal de tipo humoral, ya que es sensible a cambios mínimos en el metabolismo del hierro y tiene una corta vida media en plasma.42
Hemojuvelina
Es la más reciente de las nuevas proteínas vinculadas al metabolismo del hierro; fue descubierta en el 2004; su gen es el responsable de la hemocromatosis juvenil ligada al 1q. 43 Su descubrimiento y asociación con esta enfermedad resultan de gran importancia no solo en el diagnóstico de esta, sino para avanzar en el entendimiento de los complejos mecanismos que regulan el metabolismo del hierro.44
Aunque su función fisiológica permanece sin esclarecer, se piensa que junto al HFE y al RTf2, puede ser uno de los elementos de la cascada de señalización que controla la expresión de la hepcidina, como puede deducirse del hecho de que pacientes deficientes de HFE y HJV no manifiestan aumento de la producción de hepcidina en respuesta a la sobrecarga de hierro.38,45 La expresión de esta proteína es fundamentalmente en el hígado, el corazón y el músculo esquelético, lo que sugiere que su participación en la localización del hierro pudiera ser extendida a otros tejidos además del hígado.46
Aunque la secuencia de esta proteína no tiene rasgos característicos que expliquen su papel en el metabolismo del hierro, se plantea que es una proteína transmembrana que contiene en su estructura un anclaje GPI en su porción C- terminal , lo que implica que puede presentarse en forma soluble o asociada a células; un motivo RGD ( arginina -glicina- aspártico ) y un dominio parcial de factor von Willebrand tipo D. La presencia de los motivos RGD ha sido observada en proteínas de la superficie celular que interactúan con las integrinas durante la interacción proteína-célula y célula-célula.47
Se plantea que hay una extraordinaria heterogeneidad de alelos en la hemocromatosis debida a mutaciones en el gen HJV, pues la mayoría de las mutaciones de este gen son raras y privadas. Muchas de estas mutaciones generan codons de terminación prematura o sustituciones de aminoácidos que afectan residuos conservados de esta proteína. 48 Aunque hasta el momento hay descritas aproximadamente 24 mutaciones del gen, la más frecuente es la G 320V y más recientemente se ha descrito la Q 116X.43
Participación de las nuevas proteínas en la homeostasia del hierro
La identificación y caracterización de estas proteínas ha conducido inevitablemente a la modificación de los modelos clásicos de regulación de la homeostasia del hierro. Así, actualmente es un hecho casi indiscutible el papel central del hígado en determinar cuánto hierro se absorbe en el intestino y su influencia en la liberación del mineral a partir de los sitios de reserva. Además, hay que tener en cuenta que no se conocen mecanismos fisiológicos que regulen la pérdida de hierro, por lo que la homeostasia de este mineral dependerá de la retroalimentación que se establece entre las necesidades corporales y la absorción intestinal de hierro.49
Como sabemos, la tasa de absorción de hierro es afectada por una serie de factores que pueden actuar simultáneamente y algunos están interrelacionados entre sí. Así por ejemplo, la absorción de hierro aumenta con la disminución de las reservas corporales del mineral, el aumento de la actividad eritropoyética, la anemia o la hipoxemia, e inversamente, disminuye en presencia de la inflamación, proceso que contribuye a la anemia de los procesos crónicos.
La absorción de hierro ocurre casi exclusivamente en el duodeno y sigue una secuencia de pasos que incluyen la reducción del hierro de los alimentos de su estado férrico a ferroso, la internalización apical del mineral, el almacenamiento intracelular o el tráfico transcelular y la liberación basolateral. En todos estos eventos es indispensable la actuación de las proteínas implicadas en el metabolismo del hierro, tanto las nuevas como las tradicionalmente conocidas.
El primer paso del proceso absortivo es la reducción del hierro férrico a ferroso, que ocurre por la actividad ferrireductasa del citocromo b duodenal al nivel del borde en cepillo del enterocito (fig. 2). Una vez en estado ferroso, el hierro es transportado a través de la membrana plasmática al interior celular por el DMT 1. Dentro del enterocito, el mineral puede tener 2 destinos: ser almacenado como ferritina y excretado en las heces cuando se produce la decamación de los enterocitos senescentes, o puede ser transferido a través de la membrana basolateral al plasma a través de la ferroportina, proceso en el que se requiere la actividad ferrooxidasa de la hefastina (fig. 2). La expresión de cada uno de los genes involucrados en estos pasos está sujeta a regulación, y los cambios en su expresión han sido examinados en diferentes condiciones clínicas; entre las que la mejor caracterizada es la anemia por deficiencia de hierro, en la que se ha observado la elevación de los niveles de ARNm y de proteínas del DMT 1, del citocromo b duodenal y de la ferroportina.49

Fig. 2. Participación de las nuevas proteínas en la homeostasia del hierro.
En enterocito duodenal el Fe 3+ de la dieta es reducido a Fe 2+ por la óxido- reductasa férrica citocromo b duodenal
(itbD), internalizado por el transportador de metales divalentes (DMT1), oxidado por la hefastina y liberado a la circulación por la ferroportina ( Fpn ). En los macrófagos, el hierro liberado en la lisis de los eritrocitos senescentes es almacenado como ferritina u oxidado y exportado a la ciculación a través de la ferroportina . El hepatocito toma el hierro de la circulación como hierro libre o unido a la transferrina a través de los receptores de transferrina ( TfR1 y TfR2 ). El TfR2 sirve como sensor del hierro circulante unido a la transferrina y modula la expresión de la hepcidina . La expresión de la hepcidina es también modulada por el HFE y la hemojuvelina (HJV). La hepcidina es secretada a la circulación donde disminuye la liberación del hierro mediada por la Fpn en los enterocitos , macrófagos y hepatocitos (líneas discontinuas). ( Tomado de: Fleming RE, Bacon BR.) 49
Una vez liberado a la circulación, el hierro se une con la transferrina y es transportado a los sitios de utilización y almacenamiento. La producción de hemoglobina por el eritrón es el principal consumidor de hierro, de ahí la mayor expresión del RTf en los precursores eritroides, lo que asegura el suministro de hierro necesario. Este hierro hemoglobínico está en un recambio continuo, pues los eritrocitos senescentes son fagocitados por los macrófagos del SRE (fig. 2), donde son lisados por los fagosomas. El hierro que es liberado al citoplasma del macrófago, por mecanismos aún desconocidos, es exportado por la ferroportina, asistida por la actividad ferrooxidasa de la ceruloplasmina plasmática. Los macrófagos pueden también incorporar el hierro de la transferrina, transportarlo a través de la membrana endosomal vía DMT 1 e incorporarlo en ferroproteínas como la ferritina.32,40
Por su parte, los hepatocitos toman el hierro circulante de la sangre portal, bien sea como hierro libre o unido con la transferrina, lo almacenan como ferritina y lo liberan vía ferroportina cuando hay aumento de las demandas del mineral (fig. 2).49
Teniendo en cuenta la relevancia de la ferroportina en la liberación o exportación de hierro a partir de los enterocitos, los macrófagos y los hepatocitos, se ha planteado que esta proteína es un elemento determinante de la homeostasia del hierro.49
El descubrimiento de la hepcidina reveló la importancia del hígado en el monitoreo del estado de hierro corporal y en la modulación de la liberación del hierro celular mediada por la ferroportina, pues la hepcidina disminuye la actividad funcional de la ferroportina mediante la unión directa a esta proteína, que conduce a la internalización y degradación de la ferroportina.42 En el enterocito, esta acción podría disminuir el transporte basolateral del hierro y así disminuir la absorción del mineral. Por su parte, en los macrófagos y hepatocitos, la hepcidina puede guiar a la exportación del hierro y a la disminución de las reservas del mineral.49
La expresión de la hepcidina en el hígado es regulada por los mismos factores que afectan la absorción del hierro, por lo tanto, cuando alguno de estos factores se altera, la absorción de hierro varía inversamente con la expresión hepática de la hepcidina.49 Además la expresión de esta proteína es afectada por el RTf 2, la HFE y la HJV. Se ha planteado que estas moléculas operan por encima de la hepcidina,48 y que la ferroportina está por debajo de esta molécula en la ruta de control de la homeostasia del hierro.50
Actualmente se plantea que el hepatocito no solo es el sitio de almacenamiento de los depósitos de hierro, sino que es el centro de control del mantenimiento de la homeostasia de este mineral, pues él recibe múltiples señales relacionadas con el balance del hierro y es el responsable de del control transcripcional de la hepcidina.51
Las alteraciones en la regulación de la hepcidina han sido estudiadas fundamentalmente en 2 condiciones clínicas: la hemocromatosis hereditaria y la anemia de los procesos crónicos. Las variaciones de la homeostasia del hierro observadas en estas entidades son contrarias. Así, la expresión de la hepcidina es extremadamente baja en pacientes con hemocromatosis , mientras está aumentada en pacientes con estados inflamatorios. En la hemocromatosis hereditaria hay un incremento de la absorción del hierro dietético, una escasez relativa de hierro en los macrófagos del SRE y un aumento de la saturación de la transferrina circulante, lo que hace que los hepatocitos se carguen con hierro probablemente porque la incorporación de hierro de la circulación excede la exportación mediada por la ferroportina . Por el contrario, en la anemia de los procesos crónicos, la retención de hierro por los enterocitos duodenales y los macrófagos conduce a una saturación de la transferrina marcadamente disminuida, a una eritropoyesis restringida de hierro y a una anemia de ligera a moderada. Así, la hepcidina ofrece una explicación unificada para las alteraciones del metabolismo del hierro observadas en estas 2 condiciones clínicas.49
Indiscutiblemente, en los últimos años se han producido notables avances en la comprensión de los mecanismos que regulan la homeostasia del hierro. Sin embargo, existe consenso en que aún quedan muchas interrogantes acerca de cómo son reguladas la absorción y distribución del hierro. Algunas de ellas son: cuál es el mecanismo molecular por el que HFE, RTf 2 y HJV influyen sobre la expresión de la hepcidina ; qué influencia tiene la expresión de HFE en otras células sobre la homeostasia del hierro; qué otros genes están involucrados en el metabolismo de este mineral, como el transportador del hemo o las proteínas participantes en el tráfico intracelular; y más futurista aún, el posible potencial terapéutico para los antagonistas de la hepcidina en el tratamiento de la anemia de los procesos crónicos o la posible utilización de hepcidina exógena en el tratamiento de la hemocromatosis hereditaria.
Agradecimientos
Este artículo se elaboró dentro del programa del diplomado sobre Balance antioxidante/ pro-oxidante: salud y enfermedad, organizado por el Instituto de Farmacia y Alimentos de la Universidad de La Habana. Los autores desean agradecer al colectivo de profesores por sus valiosas orientaciones.
Summary
Iron is a very important mineral for the organism and its regulation requires a complex molecular network. Only 3 proteins that took part in iron metabolism were known a few years ago, but in the last decade, new proteins that participate in iron homeostasis and that are involved in its transportation, absorption, recycling and balance in the organism have been discovered in a sequential way, starting from the study of some genetical diseases, such as hereditary hemochromatosis. The identification and isolation of these proteins lead inevitably to the modification of the classical models of regulation of the homeostasis of this powerful mineral. A review of the esential elements known up to now of each of these new proteins and the interaction among them was made in this paper.
Key words: Iron, hepcidin, HFE, hemojuvelin.
Referencias bibliográficas
1. Roy CN, Enns CA. Iron homeostasis: new tales from the crypt. Blood 2000 ;93:4020-7.
2. Beaumont C. Molecular mechanisms of iron homeostasis. Med Sci 2004;20:68-72.
3. Cazzola M. Novel genes, proteins and inherited disorders of iron overload: iron metabolism is less boring than thought. Haematologica 2002;87:115-6.
4. Hentze MW, Kuhn LC. Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide and oxidative stress. Proc Natl Acad Sci USA 1996 ;93:8175-82.
5. Eisenstein RS, Blemings KP. Iron regulatory proteins, iron responsive elements and iron homeostasis. J Nutr 1998;128:2295-8.
6. Roy CN, Andrews NC. Recent advances in disorders of iron metabolism: Mutations, mechanisms and modifiers. Hum Mol Genet 2001;10:2181-6.
7. Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, et al. A novel MHC class I like gene is mutated in patients with hereditary haemochromatosis . Nat Genet 1996;13:399-408.
8. Feder JN, Penny DM, Irrinki A, Lee VK, Lebron JA, Watson N. The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding. Proc Natl Acad Sci USA 1998 ;95:1472 -7.
9. Gross CN, Irrinki A, Feder JN, Enns CA. Co-trafficking of HFE, a nonclassical major histocompatibility complex class I protein, with the transferrin receptor implies a role in intracellular iron regulation. J Biol Chem 1998;273:22068-74.
10. Waheed A, Grubb JH, Zhou XY, Tomatsu S, Fleming RE, Costaldi ME, et al. Regulation of transferrin-mediated uptake by HFE, the protein defective in hereditary hemochromatosis . Proc Natl Acad Sci USA 2002 ;99:3117-22.
11. Zhang AS, Davies PS, Carlson HL, Enns CA. Mechanisms of HFE-induced regulation of iron homeostasis: Insights from the W81AHFE mutation. Proc Natl Acad Sci USA 2003;100:9500-5.
12. Davies PS, Zhang AS , Anderson EL, Roy CN, Lampson MA, Mc Graw TE, et al. Evidence for the interaction of the hereditary haemochromatosis protein HFE, with the transferrin receptor in the endocytic compartments. Biochem J 2003 ;373:145 -53.
13. Pietrangelo A. Hereditary hemochromatosis - A new look at an old disease. New Engl J Med 2004 ;350:2383 -97.
14. Fleming MD, Romano MA, Su MA Garrick LM, Garrick MD, Andrews NC. Nramp 2 is mutated in the anemic Belgrade (b) rat: Evidence of a role for Nramp 2 in endosomal iron transport. Proc Natl Acad Sci USA 1998;95:1148 -53.
15. Fleming MD, Trenor CC III, Su MA, Foernzler D, Beier DR, Dietrich WF, Andrews NC. Microcityc anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet 1997;16:383 -6.
16. Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, et al. Cloning and characterization of a mammalian proton-coupled metal-ion transporter . Nature 1997 ;388:482 -8.
17. Andrews NC. Disorders of iron metabolism. N Engl J Med 1999;341:1986 -95.
18. Camaschella C. Why do humans need two types of transferrin receptor? Lessons from a rare genetic disorder. Haematologica 2005;90:296 .
19. West AP, Bennett MJ, Sellers VM, Andrews NC, Enns CA, Bjorkman PJ. Comparison of the interactions of transferrin receptor and transferrin receptor 2 with transferrin and the hereditary hemochromatosis protein HFE. J Biol Chem 2000 ;275:38135-8.
20. Fleming RE, Ahmann JR, Migas MC, Waheed A, Phillip H, Kawabata H, et al. Targeted mutagenesis of the murine transferrin receptor 2 gene produces hemochromatosis . Proc Natl Acad Sci USA 2002 ;99:10653-8.
21. Kallia P, Samara M, Stamatopoulos K, Belessi C, Stavroyanni N, Tsompanakou A, et al. Molecular evidence for transferrin receptor 2 expression in all FAB subtypes of acute myeloid leukemia . Leuk Res 2003 ;27:1101-3.
22. Wallace DF, Summerville L, Lusby PE , Subrammaiam VN. First phenotypic description of transferrin receptor 2 knockout mouse and the role of hepcidin. Gut 2005;54: 980-6.
23. Camaschella C, Roetto A, Cali A, De Gobbi M, Garozzo G, Corella M, et al. The gene TfR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat Genet 2000;25:14 -5.
24. Roetto A, Daraio F, Alberti F, Porporato P, Cali A, De Gobbi M, Camaschella C. Hemochromatosis due to mutations in transferrin receptor 2. Blood Cell Mol Dis 2002 ;29:465 -70.
25. Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libiana N, et al. Hephaestin , a ceruloplasmin homologue in intestinal iron transport, is defective in the sla mouse. Nat Genet 1999;21:195-9.
26. Levy JE, Montross LK, Cohen DE, Fleming MD, Andrews NC. The C282Y mutation causing hereditary hemochromatosis does not produce a null allele. Blood 1999;94:9-11.
27. Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in the intracellular iron metabolism. J Biol Chem 2000;275:19906-12.
28. Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, et al. Positional cloning of zebrafish ferroportin 1 identifies a conserved vertebrate iron exporter. Nature 2000;403:776-81.
29. Mckie AT, Marcioni P, Ralfs A, Brennan K, Wehr K, Barrow D, et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell 2000;5:299-309.
30. Beaumont C, Canonne-Hergaux F. Erythrophagocytosis and recycling of heme iron in normal and pathological conditions, regulation by hepcidin . Transfus Clin Biol 2005;12:123 -30.
31. Montosi G, Donovan A, Totaro A, Garuti C, Pignotti E,Cassanelli S, et al. Autosomal -dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J Clin Invest 2001;108:619 -23.
32. Fleming RE, Sly WS. Ferroportin mutation in autosomal dominant hemochromatosis loss of function, gain in understanding. J Clin Invest 2001 ;108:521 -2.
33. De Domenico I, Mc Vey D,Nemeth E, Vaughn MB, Musci G, Ganz T, Kaplan J. The molecular basis of ferroportin-linked hemochromatosis . Proc Natl Acad Sci USA2005 ;102:8955-60.
34. Forrellat M, Fernández N. Hepcidina : nueva molécula, nuevos horizontes. Rev Cubana Hematol Inmunol Hemoter 2004; 20(3). Disponible en: http://bvs.sld.cu/revistas/hih/vol20_3_04/hih03304.htm
35. Pigeon C, Ilyin G, Courseland B, Leroyer P,Turlin B, Brissot P, Loreal O. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin , is overexpressed during iron overload. J Biol Chem 2001;276:7811-9.
36. Nicolas G, Chauvet C, Viatte L, Danan JL, Brigard X, Devaux I, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia , hypoxiaand inflammation. J Clin Invest 2002 ;110:1037-44.
37. Dallalio G, Fleury T, Means RT. Serum hepcidin in clinical specimens. Br J Haematol 2003;122:996-1000.
38. Gehrke SG, KulaksezH , Hemann T, Riedel HD, Bents K, Veltkamp C, Stremmel W. Expression of hepcidin in hereditary hemochromatosis: evidence for a regulation in response to the serum transferrin saturation and to non- transferrin -bound iron. Blood 2003;102:6371-6.
39. Roy CN, Weinstein DA, Andrews NC. 2002 E. Mead Johnson Award for Reasearch in Pediatrics Lecture: The molecular biology of the anemia of chronic disease: A hypothesis. Pediatr Res 2003 ;53:507-12.
40. Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003;103:783-8.
41. Nemeth E, Tuttle MS, Pwelson J, Vaughn MB, Donovan A, Ward D, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004;306:2090-3.
42. Ajioka R, Kushner J. Another link in the chain. Blood 2004;103:2439-40.
43. Daraio F, Ryan E, Gleeson F, Roetto, Crowe J, Camaschella C. Juvenil hemochromatosis due to G320V/Q116Vcompoundheterozygosity of hemojuvelin in an Irish patient. Blood Cells Mol Dis 2005;35:174-6.
44. Celec P. Hemojuvelin: A supposed role in iron metabolism one year after its discovery. J Mol Med 2005;83:521-5.
45. Nemeth E, Valore EV, Terreto M, Sciller G, Lichtenstein A, Ganz T. Hepcidin , a putativemediator of anemia of inflammation, is a type II acute-phase protein. Blood 2003;101:2461-3.
46. Rodríguez Martínez A, Niemela O, Parkkila S. Hepatic and extrahepatic expression of the new iron regulatory protein hemojuvelin . Haematologica 2004 ;89:1441-5.
47. Giancatti FG, Ruoslahti E. Integrin signaling . Science 1999;285:102 -32.
48. Robson KJH, Merryweather -Clarke AT, Cadet E, Viprakasit V, Zaahl MG, Pointon JJ. Recent advances in understanding hemochromatosis : A transition state. J Med Genet 2004;41:721-30.
49. Fleming RE, Bacon BR. Orchestration of iron homeostasis. N Engl J Med 2005 ;352:1741-4.
50. Yeh KY, Yeh M, Glass J. Hepcidin regulation of ferroportin 1 expression in the liver and intestine of the rat. Am J Physiol Gastrointest Liver Physiol 2004;2:6385 -94.
51. Roy CN, Andrews NC. Anemia of inflammation: The hepcidin link. Curr Opin Hematol 2005;12:107-11.
Recibido: 10 de diciembre de 2005. Aprobado: 15 de diciembre de 2005
MsC. Mariela Forrellat Barrios . Instituto de Hematología e Inmunología. Apartado Postal 8070, Ciudad de La Habana, CP 10800, Cuba. Tel (537) 578268, 578695, 544214. Fax (537) 442334. e -mail: ihidir@hemato.sld.cu

