










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter v.22 n.3 Ciudad de la Habana sep.-dic. 2006
Instituto de Hematología e Inmunología
Aspectos inmunológicos del trasplante de células progenitoras hematopoyéticas
Dr. Juan Carlos Jaime Fagundo,1 Dra. Elvira Dorticós Balea,1 Dra. Valia Pavón Morán,1 Dr. Ariel Jesús Jauma Rojo2 y Dr. Lázaro Cortina Rosales1
Resumen
Los pacientes sometidos a trasplante de células hematopoyéticas experimentan un período prolongado de disfunción inmunológica que puede persistir por un año o más, con una afectación tanto de la inmunidad celular como humoral. Esto hace que tengan un riesgo elevado de infecciones en el período postrasplante. Después del acondicionamiento, hay una pérdida de la memoria de inmunidad acumulada durante toda la vida a la exposición a agentes infecciosos, ambientales y vacunaciones. El tratamiento de las infecciones se hace difícil, dado la pobre respuesta inmunitaria presente. Una de las herramientas para limitar el riesgo de infección es el uso de vacunas, aunque su efectividad se ve limitada por la inmunidad disfuncional que caracteriza a este período.
Palabras clave: reconstitución inmune, vacunación postrasplante.
Los pacientes sometidos a un trasplante de células progenitoras hematopoyéticas (TCPH), experimentan un período prolongado de disfunción inmunológica, que puede persistir por varios años.1,2 Presentan un patrón predecible de deficiencia y recuperación del sistema inmune y existe una afectación de la inmunidad tanto celular como humoral (tabla 1), lo cual hace que estos pacientes tengan un riesgo elevado de infecciones, con alta morbiletalidad. El régimen de acondicionamiento (RA), destruye la hematopoyesis normal con daño de neutrófilos, monocitos y macrófagos, así como a las células de las mucosas, y causa una pérdida temporal de la integridad de esta barrera. El tracto gastrointestinal que normalmente contiene bacterias y hongos, se convierte en un reservorio patógeno potencial.
Tabla 1. Repercusión de la Inmunidad celular y humoral en el trasplante de células hematopoyéticas
| Fenotipo | Células B | Células T | Células NK |
| Incremento de expresión de CD 10+ en médula por un año | Inversión de CD4/CD8 por 6-12 meses | Recuperación de las células CD3-CD3+/ CD56+ a niveles normales o aumentados en el postrasplante inmediato | |
| Función in vitro | Disminución de la respuesta proliferativa a Stafilococcus aureus Disminución de la producción de IgM en respuesta a antígenos
| Disminución de la respuesta proliferativa a antígenos CD3, IL-2 y reacción alogénica linfocítica mixta Disminución de los linfocitos T citotóxicos al virus Epstein Barr Disminución de la producción de IL-2 |
|
| Función in vivo | Disminución de los niveles de IgM, IgG e IgA por 3 meses, 6-9 meses y más de 2 años, respectivamente | Disminución de la hipersensibilidad retardada |
|
La recuperación de la respuesta inmune también está en dependencia del tipo de trasplante que se realice, ya que en alogénico, se adicionan otros factores (tabla 2). La recuperación de la inmunidad celular y humoral después de la exposición a altas dosis de terapia citotóxica, está en dependencia predominantemente de la reconstitución de los elementos linfoides. En el trasplante alogénico, la reconstitución inmunológica es particularmente más demorada debido a la re-educación de las células linfoides del donante en un ambiente extraño en ausencia de timo funcional. La naturaleza de la inmunidad disfuncional surge de las alteraciones cualitativas y cuantitativas de los linfocitos T y B, lo que en ausencia de enfermedad injerto contra huésped crónica (EICH-C), se resuelve en un período de 6-12 meses.3
Tabla 2. Factores que afectan la recuperación inmune después del trasplante alogénico
| Factor | Efecto |
| Fuente de células utilizada | La SP tiene una recuperación más rápida que la MO |
| Inmunosupresión para prevenir o tratar la EICH | Daño del EICL y respuesta anti-viral |
| EICH | Inmunodeficiencia, daño tímico demora la tolerancia |
| Edad del receptor (edad del timo) | Pacientes jóvenes favorecen una rápida recuperación tímica |
| Cantidad de linfocitos T | Mayor cantidad mejoran la recuperación |
| Profunda inmunoablación del receptor | Favorece la expansión de células T |
| Inmunodominancia | Favorece la expansión de células T |
| Células T reguladoras | Reduce EICH, retiene el EICL |
| Balance Th1/Th2 | Reduce EICH, retiene el EICL |
| Factores de crecimiento IL-2, IL-12, IL-7 | Favorecen la expansión de células T y la recuperación tímica |
| Quimerismo linfoide | El quimerismo mixto reduce el EICL y la EICH |
| Incompatibilidad HLA | La alorreactividad de las células favorece el EICL y la EICH |
SP: sangre periférica; MO: médula ósea; EICH: enfermedad injerto contra huésped; EICL: efecto injerto contra leucemia; IL: interleucina; NK: célula asesina natural; Th: célula T auxiliadora.
Después del TCPH, hay una pérdida de la memoria de la inmunidad acumulada durante toda la vida, a la exposición a agentes infecciosos, ambientales y vacunaciones. Debido a que la transferencia de inmunidad del donante al receptor es variable e influenciada por el tiempo de exposición antigénica entre el donante y el receptor, la inmunidad adquirida pasiva no es confiable para producir inmunidad a largo plazo.2
Reconstitución de la inmunidad celular
Después del TCPH, el número de linfocitos T y B circulantes, recupera sus valores normales en las primeras 6 semanas, manteniendo un déficit funcional durante el primer año.
La recuperación inmunológica es más rápida en los trasplantes singénicos y autólogos que en los alogénicos y en los de sangre periférica (SP), en comparación con los de médula ósea (MO).4 El número de células T CD3+ se recupera en corto tiempo después del trasplante, y más aún cuando se infunde un mayor número de células CD34. También se mantiene una inversión prolongada del índice CD4/CD8, debido a una disminución de las CD4 auxiliadoras y un aumento de las CD8 supresoras.5 Existe una deficiencia en el número de células CD45RA+, generadas durante este período y la relación CD4/CD45RA+ puede no recuperarse hasta los 2 años. Durante este tiempo, también hay una pérdida de receptores T, lo que persiste por más de un año.6
La función de las células T, se mantiene alterada durante los 6-12 meses que siguen al TCPH, y en el alogénico, el repertorio inmune es dominado por células T derivadas del donante, predominantemente células de memoria efectoras.7 La producción de interleucina 2 (IL-2) por las células T, está disminuida en respuesta a estímulos con mitógenos y las reacciones de hipersensibilidad retardada están ausentes, lo que se recupera solo cuando no existe EICH-C. La actividad citolítica de las células CD8+ está comprometida, como se demuestra en la respuesta inefectiva frente al virus Epstein-Barr (VEB).8
La fase final de la recuperación inmune celular, está dada por la emergencia de nuevas células T generadas de precursores pre-tímicos del donante en el caso del trasplante alogénico. Estas células se procesan por el tejido tímico del receptor y las hace tolerantes al alo ambiente. Existen diferencias en esta "educación" de los linfocitos según la edad del receptor. Los niños y pacientes jóvenes tienen un timo más funcional y por lo tanto, una mayor recuperación del número de células T en los primeros 2 años.9 Estudios demuestran que los pacientes que son sometidos a TCHP antes de los 18 años de edad, tienen un conteo de células CD4+ igual a los donantes pocos años después del trasplante, mientras que a los que se les realizó el proceder después de esta edad, mantenían el conteo más bajo. Esto sugiere que en pacientes mayores de 18 años la insuficiencia tímica pos trasplante no desaparece completamente.10
En contraste, la reconstitución de las células asesinas naturales (NK) no requiere un timo funcional y ocurre rápidamente en las primeras semanas del período postrasplante.1,10
El número de células CD3- CD16+ CD56+ está aumentado en el primer mes del trasplante alogénico y autólogo, e incrementan sus niveles en los primeros 3 meses.
Los neutrófilos se recuperan en 2 ó 3 semanas, pero se mantiene una disminución de la quimiotaxis por un período de hasta 4 meses. El número de monocitos en la SP regresa a los valores normales en los primeros 40 días y su función es generalmente normal. Los macrófagos aparecen en el hígado y en los pulmones cerca del día 80 y se ha demostrado que son del donante.10
Reconstitución de la inmunidad humoral
En el período postrasplante inmediato, las células B medulares expresan el antígeno temprano CD10. Durante el primer año del trasplante, estas células expresan CD1c+, CD5+, CD23+ y CD38+, así como IgM e IgD unidos a la membrana. En ausencia de EICH, las células periféricas que expresan CD19+ y CD20+, regresan a la normalidad después de 3-6 meses postrasplante, y la recuperación es más rápida en pacientes que reciben trasplante autólogo de sangre periférica, dado el aumento de células T auxiliadoras CD4+/CD45RO+.11 La respuesta proliferativa de células B a mitógenos, está casi abolida durante los primeros meses. Las concentraciones séricas de IgM son normales en los primeros 3 a 6 meses postrasplante, pero las de IgG se mantienen muy bajas durante al menos 9 meses, y los niveles de IgA pueden demorar hasta 2 años en recuperarse.1,12
La ausencia de células T auxiliadoras para la estimulación de las células B, resulta en una dificultad para el cambio de clase de inmunoglobulina de IgM a IgG, después de la exposición antigénica. La recuperación de IgG 2 y IgG 4, está particularmente demorada, y en los primeros 3 meses del trasplante los pacientes no logran incrementar los niveles de IgM primaria en respuesta a la inmunización.12
En general, la recuperación inmunológica depende de un número de factores (tabla 3) y se produce más rápidamente en los trasplantes de SP que en los de MO, ya que el primero contiene 10 veces más células T y B y existe un aumento fundamentalmente de células CD4+, así como una relación CD4/CD8 más alta que en los de MO.4 También se han observado diversos fenómenos auto inmunes (anemia hemolítica, trombocitopenia), así como la transmisión desde el donante al receptor de enfermedades alérgicas (asma, hipersensibilidad) y auto inmunes (tiroiditis, diabetes).13,14
Tabla 3. Factores que afectan la recuperación inmunológica, según el tipo de trasplante
| Tipo de trasplante | Factor predisponente |
| Autólogo |
|
| Alogénico | Además de los factores presentes en los autólogos :
|
EICH: enfermedad injerto contra huésped.
Debido al daño inmunológico antes descrito, los pacientes experimentan infecciones en diferentes períodos postrasplante, lo que refleja el defecto predominante en el huésped (figura). Esto hace que existan patrones de infecciones en este período, esquematizados en 3 fases, que comienza desde el día 0 del trasplante.15
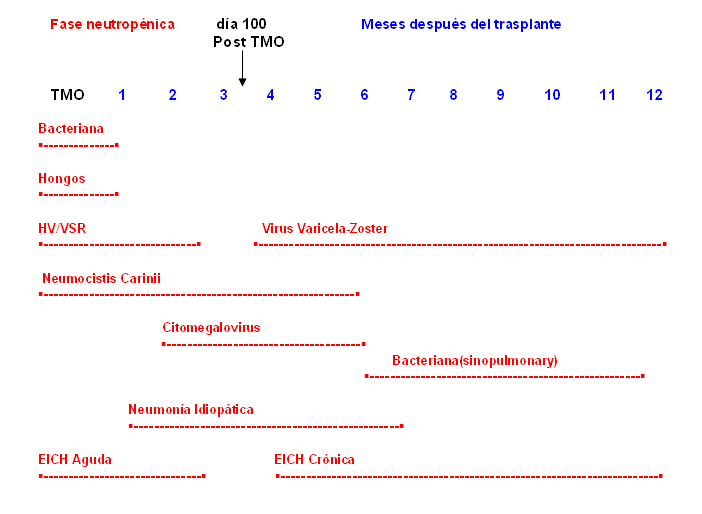
Figura: Patrones de infección que siguen al trasplante de médula, que muestra el tiempo aproximado de la aparición de las complicaciones.
TMO: trasplante de médula ósea; EICH: enfermedad injerto contra huésped; HV: herpes virus;
VSR: virus sincitial respiratorio.
Fase I, pre implante. Desde el día 0 hasta el día 30 del trasplante (primer mes). En este período los pacientes tienen 2 factores de riesgo para la infección, que son la neutropenia mantenida y la ruptura de la barrera cutáneo-mucosa. Generalmente los primeros episodios febriles son de causa bacteriana y solo en ocasiones se puede identificar un germen o sitio de infección, por lo que estos cuadros se tratan de forma empírica. Los agentes que prevalecen son grampositivos, la Candida y si la neutropenia continúa, el Aspergillus. En ocasiones puede haber reactivación del virus herpes simple.
Fase II, posimplante. Esta fase comprende desde el día 30 hasta el día 100 y se caracteriza por el daño de la inmunidad mediado por células; la duración de este defecto se puede prolongar por la presencia de la EICH y la terapia inmunosupresora utilizada para esto. Después del implante, los herpes virus y particularmente el CMV son los agentes patógenos más frecuentes y puede causar cuadro de neumonía, hepatitis y colitis. También son frecuentes durante este período los gérmenes gramnegativos, el Pneumocystis carinii y algunas especies de Aspergillus.
Fase III, Fase tardía. Las infecciones son más frecuentes en los pacientes con trasplante alogénico que sufren de EICH. En estos casos se mantienen los defectos de inmunidad humoral y celular y daño en la función del sistema reticuloendotelial, y los pacientes tienen riesgo de sufrir infecciones por CMV, virus varicela-zóster, virus Epstein-Barr, virus respiratorios y por bacterias encapsuladas como el H. influenzae y el S. pneumoniae.
Aspectos de la respuesta aloinmune
La aloinmunidad es un proceso complejo, que involucra tanto a los linfocitos T como a las células NK del donante, que interactúan con células específicas del receptor.
Células T: existen varias fases en la alorrespuesta. En la fase inicial o de inducción, las células CD8+ y CD4+ del donante, interactúan con antígenos del complejo mayor de histocompatibilidad (CMH), clase I y clase II respectivamente, que se encuentran en las células presentadoras de antígenos (CPA) del receptor. La señal dada por el CMH a través de los receptores de células T y de las células CD3+, CD4+ ó CD8+, constituyen la primera señal para la activación de las células T. Una respuesta completa de las células T, lleva a la proliferación de las células efectoras, requiriendo una segunda señal dada por la interacción con moléculas co-estimuladoras tales como CD80 y CD86 sobre las CPA y las células CD28+ en la superficie de los linfocitos. En esta segunda fase o de de expansión, las células T proliferan bajo la influencia de la IL2 e IL12. En la tercera fase o efectora, las células T aloactivadas interactúan con sus antígenos similares en las células dianas del receptor, causando daño celular por citotoxicidad directa mediante la liberación de perforina y granzima, o por la producción de citocinas inflamatorias.16
Células NK: mientras que las células T requieren una estimulación y posterior expansión, la interacción de las células NK con otras células, produce una señal positiva para destruir inmediatamente el blanco mediante la liberación de perforina y granzima. Normalmente estas células están inhibidas por señales negativas a través de los receptores KIR, que interactúan con las moléculas clase I del sistema HLA de las células diana. Cuando se encuentran con células que tienen pérdida de las moléculas clase I ó expresan estas moléculas y no son reconocidas como propias (como ocurre en los trasplantes HLA no compatibles), no se produce la señal negativa, y se liberan sustancias como la perforina y granzima.17
Las reacciones aloinmunes son responsables de 3 eventos principales, que son: el implante, la EICH y el efecto injerto contra tumor (EICT).
Implante
Tanto las células T como las NK del donante y del receptor, así como las células CD34+ del donante, están involucradas en la toma del injerto, y se necesita una importante inmunosupresión para permitir que esto ocurra. En los trasplantes HLA idénticos, el implante es el resultado de la acción de una alorrespuesta exitosa contra las células T residuales del receptor, que causan la eliminación del sistema inmune de este.
El uso de altas dosis de inmunosupresores en los trasplantes no mieloablativos con fludarabina y ciclofosfamida, muestra la importancia del injerto de las células T en el establecimiento de una hematopoyesis duradera. En las primeras semanas después de este tipo de trasplante, el 100 % de las células T son de origen del donante, mientras que la recuperación de la hematopoyesis es predominantemente del receptor. Semanas o meses más tarde, la hematopoyesis del receptor es sustituida por la del donante, seguido por lo que se considera efecto injerto contra médula del trasplante. En contraste, los receptores que no logran un injerto temprano de las células T del donante, tienen un alto riesgo de rechazo por las células residuales del receptor.18 Aunque algunas investigaciones han demostrado la importancia de la resistencia a la radioterapia de las células NK del receptor en el fallo de injerto, estas células tienen un papel limitado en este aspecto.
Tanto el acondicionamiento, como un número importante de stem cell, son capaces de sobrepasar los mecanismos de resistencia por las células NK del receptor. Las células NK del donante desempeñan un papel importante en la toma del injerto, ya que estas lo favorecen a través del reconocimiento y destrucción de los linfocitos y células hematopoyéticas residuales en el receptor.19
Es conocido que las células CD34+ influyen en el injerto de diferentes formas. Estas células pueden bloquear la función de las células T del receptor, a través de un efecto de veto que lleva a la apoptosis de las células T reactivas del receptor.20 En los trasplantes depletados de células T, las células CD34+ del donante son la fuente principal de células NK que favorecen el injerto tanto en los trasplantes HLA idénticos como no idénticos, por lo que un alto número de células CD34+ tiene un efecto beneficioso en este proceso.21,22
Enfermedad injerto contra huésped (EICH)
La EICH es la principal complicación de los TCPH alogénicos y de órganos que contienen células linfoides. En esta entidad se conjugan una serie de eventos inmunológicos entre el tejido injertado y el receptor, disparados por sus diferencias antigénicas, lo cual produce diversas manifestaciones clínicas, desde leves a muy severas, que pueden incluso comprometer la vida del paciente
La denominación inicial de EICH fue propuesta por Billingham 21 en 1966, quien enunció los siguientes criterios para la presentación de esta entidad:
- El injerto debe contener células inmunológicamente competentes.
- Antígenos tisulares del hospedero diferentes de los del donante.
- Receptor inmunocomprometido.
La EICH tiene 2 formas clínicas: aguda (EICH-A) y la crónica, con características clínicas, inmunológicas e histológicas diferentes. Clásicamente se ha definido que la aguda se presenta en los primeros 100 días postransplante, especialmente entre los días +7 a +21, mientras que la crónica es aquella que se manifiesta después de los 100 días.
La forma aguda afecta fundamentalmente el hígado, piel y tracto gastrointestinal, mientras que la crónico se comporta como una enfermedad multisistémica con grados variables de queratoconjuntivitis sicca, enfermedad hepática similar a la cirrosis biliar primaria, diarrea, bronquiolitis obliterante, polimiositis, así como neuropatía periférica, y aunque aparece después de 100 días postrasplante, en algunos pacientes pueden observarse manifestaciones tempranas, en los primeros 40 días.
Las células T transfundidas son las responsables de la inducción de la EICH-A, pues presentan una actividad citolítica y/o auxiliadora frente a las células del receptor que expresan antígenos diferentes. Las células efectoras de la respuesta inmunológica son los linfocitos T, principalmente CD4+, CD8+ y, en modelos animales, los linfocitos NK. En la piel y el tracto gastrointestinal estas células efectoras producen daño celular promoviendo la apoptosis.22 Existe una estrecha relación entre infección y EICH-A.
La fisiopatología de esta enfermedad se fundamenta en la distorsión de la respuesta celular contra infecciones por virus y bacterias gramnegativas en los principales órganos blanco, especialmente en el tracto gastrointestinal, donde estimulan la liberación de citocinas y mediadores inflamatorios. La piel, el hígado y el tracto gastrointestinal están expuestos a una gran cantidad de endotoxinas que disparan y amplifican la respuesta inflamatoria en los pacientes con EICH-A
La fisiopatología de la EICH-A se divide en 3 fases: el régimen condicionante, la activación de células T y la fase efectora.
Primera fase: régimen condicionante:
La EICH-A comienza desde la fase de preparación pretrasplante, es decir, antes de la infusión de las células del donante. Durante este período, se producen daños en la mucosa intestinal, el hígado y otros tejidos por la quimioterapia y la radioterapia. Las células activadas secretan citocinas proinflamatorias como el factor de necrosis tumoral a (FNTa), la IL-1 y factores de crecimiento como el factor estimulador de colonias granulomonocítico (FEC-GM), promoviendo el aumento en la expresión de moléculas de adhesión y complejos mayores de histocompatibilidad.23
Segunda fase: activación de las células T del donante:
Esta fase incluye la presentación antigénica, la activación de los linfocitos T del injerto y la proliferación y diferenciación de estas células activadas. Cuando las células CD4+ y CD8+ ingresan al torrente sanguíneo, interactúan con las células presentadoras de antígeno; si existen diferencias en el CMH, las células del injerto desencadenan una reacción injerto contra hospedero. Cuando el donante y el receptor son HLA idénticos, puede presentarse EICH por diferencias en los complejos menores de histocompatibilidad. En el lecho capilar, las células T del injerto interactúan con las células endoteliales y se exponen a nuevos aloantígenos. En un proceso dinámico, el endotelio es activado y participa a su vez en la activación de las células T.23
El aumento de la E-selectina, una molécula de adhesión endotelial, y diferencias en las isoformas de PECAM (molécula de adhesión endotelial plaquetaria) entre donante y receptor, incrementan el riesgo de EICH-A. Estas moléculas permiten la adhesión de linfocitos circulantes provenientes del donante a diferentes antígenos endoteliales del huésped, lo que facilita el paso de las células a los diferentes tejidos.
En la activación de las células T se requiere de 2 señales: la interacción del TCR con péptidos unidos al HLA y las señales co-estimuladoras con las células presentadoras de antígeno. Las células T activadas secretan citocinas tipo Th1, especialmente IL-2 e IFN-g. La IL-2 es producida por las células CD4+ del injerto en los primeros días pos TMO y posteriormente por las células del hospedero. El IFNg es muy importante en la patogénesis de la EICH porque estimula la producción de múltiples citocinas en los macrófagos, amplificando de esta manera la cascada de eventos inmunológicos. 23,24 El balance entre citocinas Th1 y Th2 es crítico para la presentación de la EICH; el incremento de Th2 como de la IL-4 se relaciona con baja frecuencia y prevención de esta entidad. El uso de factor estimulador de colonias granulocítico (FEC-G) disminuye el riesgo de EICH y mejora la toma del injerto al cambiar el perfil de citocinas por aumento de las tipo Th2.23
Tercera fase: fase efectora de la inflamación:
En esta fase, los linfocitos grandes granulares (células NK) son responsables de los efectos citolíticos que se incrementan por los mediadores inflamatorios y las citocinas. La respuesta inflamatoria amplifica el daño celular al estimular la síntesis de FNT-a, IL-1a e incrementar las moléculas HLA clase II y de adhesión ICAM-1, en la piel y el tracto digestivo.23,24
Durante la fase del régimen condicionante, pueden producirse grandes cantidades de LPS (endotoxina) en la piel y el tracto gastrointestinal; en la fase efectora, el LPS estimula en la piel a los queratinocitos y en la dermis y el tracto gastrointestinal a los fagocitos mononucleares para la producción de FNTa, que produce daño directo del tejido al inducir la muerte celular por apoptosis. Los receptores TLR han sido identificados como receptores para el LPS. Algunas mutaciones de estos receptores están en relación con una pobre respuesta a estas toxinas y se reduce el riesgo de EICH aguda.25 Los agentes virales también agravan la EICH y son factores predisponentes para su presentación; entre estos se destaca la infección por citomegalovirus, el cual estimula el fenómeno inflamatorio y la producción de citocinas tipo Th1.23 La citotoxicidad es mediada por las células NK y depende de las vías de Fas-L, perforinas y granzimas, asociándose con el daño celular en los órganos afectados.23
La EICH-C es similar a las enfermedades autoinmunes como la esclerodermia; histológicamente se encuentra un notorio aumento del depósito de colágeno y fibrosis en los órganos afectados, asociado con una alta producción de autoanticuerpos. Las células T del donante reconocen antígenos específicos del hospedero en la piel y la sangre que desencadenan fenómenos de alorreactividad y autorreactividad. Se ha demostrado la aparición de anemia hemolítica, trombocitopenia, anticuerpos antilinfocitos y autoanticuerpos contra el núcleo, nucleolo, músculo liso, tiroides y piel.24
Los estudios de inmunotipificación de linfocitos han mostrado infiltrados de células T (la mayoría son CD8+), sin NK o linfocitos B en la piel. Se han encontrado altos niveles de IFN-g, lo que sugiere un perfil de citocinas Th1. Además, se ha demostrado la presencia de FNT-a e IL1-a en los queratinocitos de lesiones de EICH-C, lo que amplifica el fenómeno inflamatorio. La presencia de cambios esclerodermiformes está relacionada con la proliferación de los fibroblastos y aumento en la producción y depósito de colágeno, asociados con clones CD4+ autorreactivos y con la degranulación de mastocitos.24
Efecto injerto contra tumor (EICT)
En el TCPH alogénico, se ha reconocido que existen mecanismos inmunológicos mediados por las células inmunes del donante, que tienen un papel importante en la erradicación de células malignas. Por lo tanto, se considera que el potencial curativo en el trasplante alogénico no es solo dependiente del régimen de acondicionamiento, sino también del llamado efecto injerto contra tumor (EICT), y se piensa que es la principal razón por la que el trasplante alogénico tiene menor porcentaje de recaída que el autólogo, en los cuales se han usado los mismos regímenes de acondicionamiento.25
El conocimiento de este mecanismo ha llevado al desarrollo de procederes en la trasplantología moderna, como son la infusión de linfocitos del donante (ILD) y el trasplante no mieloablativo.
Las células efectoras involucradas en el EICT, incluyen diferentes subtipos del donante, tales como: células NK, células T tumor específica o asociadas con antígenos tumorales y células T específicas para antígenos de incompatibilidad menor del receptor.26,27
Las células malignas del receptor presentan antígenos que son capaces de producir respuesta y proliferación de los linfocitos T del donante, y tales líneas leucémicas específicas de células T se han utilizado con éxito en el tratamiento de la leucemia mediante la llamada transferencia o terapia adoptiva.
Este EICT no está bien comprendido. En el caso de la LMC, hay evidencia que tanto las células CD4+ cono las CD8+, están involucradas en este efecto, y estos linfocitos citotóxicos inhiben la formación del clon leucémico. Estas líneas pueden mostrar citotoxicidad solo a las células leucémicas, a estimuladores no leucémicos o a ambas, y sugiere la presencia tanto de antígenos específicos de línea y compartidos.28
El EICT puede estar mediado tanto por antígenos de compatibilidad menor (AgHm), los que dependiendo de la especificidad de línea pueden o no causar también EICH, así como por antígenos específicos tumorales, los cuales no inducen EICH.29
El mecanismo efector utilizado por los linfocitos T del donante para producir el EICT, incluye la actividad citotóxica directa a través del ligando Fas Fas L (CD95- CD178), a través de la perforina y granzima, así como mediante la secreción de citocinas, las cuales pueden lisar las células dianas, como el FNT a.
Los antígenos diana del EICT, no están bien definidos, y la estrecha relación entre este efecto y la EICH, hace pensar que se trate de antígenos compartidos entre las células malignas y tejidos normales como piel, hígado e intestino. En el caso de los trasplantes compatibles, parece estar en relación con los antígenos del sistema menor de histocompatibilidad, algunos de los cuales parecen ser tejido específicos, lo que puede contribuir a la explicación del EICT sin EICH.29
Recientes investigaciones sugieren la existencia de proteínas expresada en las células tumorales, como resultado de translocaciones o mutaciones, así como de proteínas normales que son sobre expresadas como consecuencia de una diferenciación aberrante, las que son procesadas a péptidos y presentadas en la superficie celular, y son entonces reconocidas por las células T. Estas proteínas involucradas en la transformación celular tales como el gen ras o bcr/abl, son blancos específicos tumorales para la inmunoterapia.30 De igual forma, se han aislado células T específicas, capaces de reconocer proteínas normales sobre expresadas en las células leucémicas, tales como la proteinasa-3, mieloperoxidasa y la proteína del tumor de Wilms (WT-1), que pueden estimular la citotoxicidad tanto autóloga como alogénica. En el caso de la proteinasa 3, está presente en los gránulos primarios mieloides y sobre expresada en la leucemia mieloide crónica (LMC) y algunos tipos de leucemias mieloide agudas (LMA).
El WT-1, está sobre expresado en las leucemias agudas, pero no así en las células hematopoyéticas normales, donde es muy bajo o no se expresa.31,32
Existen otras hemopatías malignas que son menos susceptibles a este efecto, lo cual se debe fundamentalmente a 2 causas:
- La rapidez de proliferación de la enfermedad, donde aquellas que tienen un crecimiento más lento responden mejor.
- Fenotipo leucémico, variando la sensibilidad según la patología:
- Sensibles: LMC, leucemia linfocítica crónica (LLC), linfomas no Hodgkin (LNH) de bajo grado.
- Poco sensibles: LMA, LNH de grado intermedio, mieloma múltiple (MM), HDG, hipernefroma, cáncer de mama.
- Insensibles: LLA, LNH de alto grado.
El EICT y la EICH están estrechamente ligadas, y está bien documentado que el desarrollo y severidad de la EICH es inversamente proporcional a la posibilidad de recaída en un alotrasplante.
El hecho de que el efecto injerto contra tumor desempeñe un papel decisivo en la erradicación de la malignidad, se ve sustentado por varias observaciones.
- Ha podido demostrarse la presencia de enfermedad mínima residual en la primera etapa posterior al trasplante, después haber recibido el paciente altas dosis de quimioterapia, como tratamiento condicionante, lo que desaparece en los primeros 9-12 meses postrasplante.
- Hay una disminución del riesgo de recaída en pacientes que padecieron una EICH aguda o crónica después del trasplante.
- Se ha demostrado que el riesgo de una recaída en un paciente sometido a un trasplante singénico es mayor, al igual que cuando se realiza la depleción de linfocitos T de la médula a implantar.
- La inducción de la remisión en pacientes que recayeron después de un alotrasplante con infusión de linfocitos T del donante.
- Demostración de la reactividad de los clones derivados de células T del donante contra células malignas del receptor.
- La suspensión brusca de la inmunosupresión en pacientes con recaída de la leucemia después de un trasplante alogénico puede ocasionalmente inducir una nueva remisión.
En la EICT se plantea que se trata de antígenos diferentes a los que producen la EICH, pero un número mayor de autores sugieren que la existencia de este efecto sin EICH, está dado por una mayor sensibilidad de la célula leucémica a un mecanismo inmunológico común. También se considera que el aumento de selectividad contra la célula leucémica podría resultar de la reacción con antígenos relacionados con la línea hematopoyética o antígenos leucémicos específicos, ya que de hecho se han descrito AgHm asociados con la línea hematopoyética.9,33
Varias características de la respuesta de las células T a los AgHm, pueden contribuir a su eficacia anti- tumor:
- Los AgHm son altamente inmunogénicos y las células T pueden causar EICH, a pesar de la inmunosupresión que se administra.
- La mayoría de los clones de células T específicos para AgHm, tienen una gran capacidad de reconocer antígenos tumorales.
- La reapuesta de células T a los AgHm, involucra tanto a los linfocitos CD4+, como a los CD8+ y aunque las célula leucémicas no son blanco directo de las CD4+, estas puede aumentar el EICT, aumentando y manteniendo la respuesta de las células CD8+.
Por otro lado, en los trasplantes de donantes femeninas en receptotes masculinos, existe un incremento del EICT y EICH. Esto sugiere que los AgHm, codificados por el cromosoma Y, confieren una contribución adicional a estos efectos, además de los que producen los AgHm autosómicos.26,28
El riesgo de recaída leucémica es menor en aquellos trasplantes alogénicos y no manipulados, que en los que se desarrolló le EICH aguda y crónica. Sin embargo, en pacientes que no desarrollaron esta complicación, hubo menor incidencia de recaída que en los pacientes que recibieron trasplante singénico o depletado de células T, lo que sugiere que las células T pueden actuar sobre los antígenos menores sin producir EICH.34 Una disociación del EICT de la EICH, también se ve el trasplante no mieloablativo o en la ILD, donde la regresión del tumor no coincide siempre en tiempo con el desarrollo de la EICH.35
A pesar de la existencia de innumerables tratamientos tales como esteroides, la globulina antilinfocítica (GAL), y anticuerpos monoclonales contra el FNT, CD 54 y CD 25, la EICH se mantiene como un problema importante después del TCPH.
Por esta razón, se desarrollaron técnicas para producir una depleción de células T y de esta forma prevenir la EICH hace más de 20 años, pero el entusiasmo por este proceder disminuyó rápidamente, dado el hallazgo de un incremento del rechazo, recaída y un aumento de las infecciones por una demora en la recuperación inmunológica.36 De esta forma, mientras la depleción de células T previene la muerte por EICH, compromete el EICT. Estos datos sugieren que ambos efectos están muy relacionados, y que el desarrollo de EICH, está asociado con un bajo riesgo de recaída. Sin embargo, la leucemia puede curar, en ausencia de EICH, mediante la utilización de infusión de linfocitos del donante, por lo que indica que existe una base para separar ambos fenómenos.35
En los últimos años, el reto ha sido lograr un método fiable para prevenir la EICH y mantener el EICT, así como la inmunidad celular contra agentes infecciosos.36
Es posible lograr la separación de la EICH, del EICT por diferentes métodos, tales como:
- Depleción selectiva de células T: mediante la separación de células T para antígenos de células no leucémicas, manteniendo el EICT, utilizando la purga fotodinámica. También se ha utilizado la inducción de células T, mediante inmunotoxinas contra el Ag CD25 en trasplantes haploidénticos, lo que logra un bajo porcentaje de EICH y una recuperación inmunológica superior a los casos con depleción de células T.37
- Eliminación de células T vírgenes: se ha visto que estas células, que nunca han encontrado sus antígenos específicos, expresan CD 62L+, lo que las diferencia de las células de memoria que pueden ser CD 62L- o CD 62L+. Por lo tanto, las células del donante que nunca han estado en contacto con sus aloantígenos, son las que producen la EICH, y las células activadas de memoria no inducen EICH, ya que no son estimuladas por los antígenos y sí mantienen el EICT.
- Selección de células Th2/Tc2: se han definido 4 subtipos de células T funcionales, como son: CD4+ auxiliadoras (subtipos Th1 o Th2) y células CD8+ citotóxicas (subtipos Tc1 o Tc2). Th1 y Tc1 secretan predominantemente IL2 y INFg, mientras que las Th2 y Tc2, secretan IL 4, IL5 e IL 10, que son citocinas antiinflamatorias. Por lo tanto, las células Th2 del donante no inician la EICH, y además regulan esta. Las Tc2 del donante no causan EICH importante y median el EICT, así como tienen un efecto en la prevención del rechazo al injerto. Por lo tanto, la selección de las células Th2 y Tc2, permite un injerto con preservación del EICT y prevención del EICH.38
- Inserción de genes suicidas: este efecto fue descrito por primera vez por Bordignon y colaboradores,39 y consiste en la transducción de linfocitos del donante con el gen de la enzima timidin kinasa del herpes virus. Estas células son seleccionadas e infundidas al paciente, como parte de las CPH iniciales o como ILD. Si ocurre la EICH y requiere intervención terapéutica, se administra Ganciclovir para eliminar estas células y así el EICH.
- Adición de células T reguladoras: estas células con doble marcador CD4+, CD 25+ son importantes en la inducción y mantenimiento de la autotolerancia, y producen así una disminución en la autoinmunidad.
- Uso de células NK alorreactivas para crear un EICT sin EICH.
Vacunación del paciente trasplantado
Los pacientes que son sometidos a un régimen mieloablativo y se les realiza un TCPH, experimentan un prolongado período de disfunción inmunitaria que persiste por un año o más. Los títulos de anticuerpos para enfermedades prevenibles adquiridos por vacunas declinan durante los primeros 4 años después del trasplante, si el paciente no es revacunado. Las vacunas con virus vivos atenuados son más eficaces que las que contienen virus muertos, pero las primeras son más peligrosas en pacientes inmunocomprometidos, por el riesgo de diseminación de la infección.40
Se han publicado varias guías para la inmunización de los pacientes trasplantados, aunque estas recomendaciones no han sido uniformes. En 1995 el Grupo Europeo para el Control de Enfermedades Infecciosas en Pacientes Trasplantados, publicó los resultados de un estudio realizado en Europa. La mayoría de los centros iniciaron sus vacunaciones al año después de realizado el trasplante.41
En 1997 el Centro de Control y Prevención de Enfermedades de Estados Unidos (CDC), estableció las guías para la prevención de enfermedades infecciosas en los pacientes trasplantados, que se publicó en el año 2000, junto con un esquema de inmunización en estos pacientes.42 La tabla 4 muestra las vacunas recomendados para los receptores de trasplantes hematopoyéticos.
Tabla 4. Vacunas recomendadas para receptores de trasplante de médula ósea
| Vacuna | Edad del paciente | Tiempo de administración | Comentarios |
| Difteria, tétanos, pertusis (DPT) | <7 años | 12, 14 y 24 meses después del trasplante | No hay datos relacionados con la seguridad e inmunogenicidad de la vacuna contra la tosferina, pacientes con contraindicación a esta vacuna deben recibir la DT |
| Difteria y toxoide tetánico (DT) | >7 años | 12, 14 y 24 meses después del trasplante | Pacientes con contraindicación a la pertusis deben recibir DT |
| Tétanos y toxoide diftérico (Td) | Todos los pacientes | 12, 14 y 24 meses después del trasplante | Debe ser reactivada cada 10 años |
| Haemophilus influenzae tipo B conjugada | Todos los pacientes | 12, 14 y 24 meses después del trasplante |
|
| Hepatitis B
| <18 años en pacientes susceptibles Adultos: pacientes con factores de riesgo | 12, 14 y 24 meses después del trasplante | No recomendada en todos los pacientes Se recomiendan altas dosis para adultos inmunocomprometidos, no datos relacionados con la respuesta a altas dosis en niños. Se observa una respuesta de 1-2 meses después de la tercera dosis. Los que no responden deben recibir un segundo ciclo de 3 dosis |
| Antineumocóccica | Todos los pacientes | 12 y 24 meses después del trasplante | Eficacia limitada en el postrasplante inmediato. El rango de eficacia aumenta en períodos posteriores. Se usa profilaxis antibiótica en pacientes con EICH crónica |
| Influenza
| Todos los pacientes | Vacunación antes del trasplante y reactivación a los 6 meses después del trasplante | Niños menores de 9 años deben recibir 2 dosis en la primera vacunación. Niños = 12 años deben recibir la mitad de la dosis. Los niños >12 años deben recibir la mitad de la dosis o la dosis completa |
| Antipoliomelitis
| Todos los pacientes | 12, 14 y 24 meses después del trasplante | La vacuna es inmunogénica después del trasplante, aunque no hay certeza de su eficacia |
| Parotiditis, rubéola, sarampión (PRS) | Todos los pacientes | > 24 meses después del trasplante | Se recomienda en pacientes recuperados inmunológicamente, no indicada en pacientes que reciben terapia inmunosupresoras |
| Varicela | Contraindicada | Contraindicada | Contraindicada |
Modificado del Joint Guidelines por el Centro para el control de enfermedades, Sociedad Americana de Enfermedades Infecciosas y Sociedad Americana de Trasplante de Médula Ósea, octubre 20, 2000.
En general, las respuestas a sub-unidades de proteínas en los pacientes con cáncer están muy disminuidas y la administración de vacunaciones tratando de producir respuesta son nulas en pacientes que usan Ciclosporina.43 La vacunación contra antígenos polisacáridos de gérmenes encapsulados tiene un comportamiento igual en estos pacientes, ya que las respuestas son dependientes de las células B, que son amplificadas por las células T auxiliadoras y estos aspectos inmunológicos están ausentes en el paciente postrasplantado, resultando en una respuesta inefectiva a la vacunación. Los pacientes que reciben trasplante de SP tienen una relativa preservación del título de anticuerpos en relación con los que son sometidos a TMO.44
Existen varias estrategias para mejorar la eficacia de las vacunaciones, como el uso de agentes adyuvantes y citocinas que aumentan la respuesta celular y humoral. Las sales de aluminio y el aceite mineral se han usado para mejorar la respuesta a la inmunización, pero los mecanismos de acción no han sido bien dilucidados, y se cree que inducen una reacción inflamatoria con reclutamiento de células presentadoras antigénicas y liberación de citocinas, lo que incrementa la respuesta celular y humoral.40 Dentro de las citocinas utilizadas en pacientes inmunocomprometidos para mejorar la respuesta a antígenos específicos, se encuentran la IL-1, IL-2, IL-6, IL-12 y el FNT a. 45,46 También se ha utilizado el FEC-GM, que induce la liberación de la IL-1, IL-2 y FNT a, lo que promueve la diferenciación de células T y B, así como de las CPA.47
Otras investigaciones sugieren que la presencia de títulos de anticuerpos contra enfermedades específicas en el donante influyen en el riesgo y severidad de la infección en los receptores de trasplantes alogénicos.48 Una de las estrategias para mejorar la eficacia de la vacunación postrasplante es la inmunización del donante tratando de generar inmunidad que puede pasar de forma pasiva al receptor, lo que lleva a una disminución significativa de infección por varicela zóster en receptores de donantes que han sido inmunizados 2-4 semanas antes de la obtención de las células hematopoyéticas.49 En el caso de los trasplantes autólogos, los pacientes pueden ser vacunados antes del proceder, lo que logra una respuesta más rápida a la revacunación postrasplante. Otra estrategia utilizada para prevenir enfermedades en el período postrasplante es la vacunación de los familiares allegados al paciente, así como del personal que atiende estos casos, con vacunas contra enfermedades como hepatitis, influenza, sarampión, parotiditis, rubéola y varicela. También pueden ser inmunizados de forma pasiva directamente, mediante el uso se globulinas como las de la hepatitis, rabia, sincitial respiratorio, tétanos y varicela zóster.50
A continuación se muestran algunas consideraciones sobre vacunas específicas y su manejo en los pacientes trasplantados:
- DPT, polio y hemofilus: estas son recomendadas en pacientes después del primer año. Es importante la administración de más de una dosis para facilitar la respuesta anamnésica, lo que se logra con las reactivaciones anuales.
- Pneumocóccica: aunque la respuesta inmune está dañada para los polisacáridos neumocóccicos, se logra alguna protección con el esquema de 2 dosis a los 12 y 24 meses.
- Influenza: se recomienda la vacunación anual, comenzando a los 6 meses después del trasplante, ya que la respuesta antes de este tiempo es mínima.
- Hepatitis B: no recomendada en todos los pacientes y solo en aquellos grupos de riesgo.
- Hepatitis A y anti meningócoccica: no se recomiendan de rutina. La vacuna anti meningóccica se pudiera administrar en pacientes que han sido esplenectomizados previamente, pero su uso está por confirmar.
- Parotiditis, rubéola y sarampión (PRS): el uso de virus vivos atenuados está restringido en los receptores de TCPH. Su uso no debe ser antes de los 2 años postrasplante, pero la efectividad y seguridad en su uso no han sido establecidas.
- Varicela: aunque se han obtenido buenos resultados en algunos estudios, su efectividad y seguridad no han sido establecidas para ser recomendada de forma rutinaria.
- Rabia: su administración no ha sido recomendada.
Summary
Immunological aspects of the transplant of haematopoietic progenitor cells
Patients undergoing transplant of haematopoietic cells experience a prolonged period of immunological dysfunction that may persist for a year or more, with an affectation of cellular and humoral immunity. This makes them have a high risk of infections in the posttransplant period. After the conditioning, there is a loss of memory of the immunity accumulated during the whole life to the exposure of infectious and environmental agents, and vaccines. The treatment of the infections is difficult due to the poor immunitary reponse. One of the tools to limit the risk of infection is the use of vaccines, although their effectiveness may be restricted by the dysfunctional immunity characterizing this period.
Key words: Immune reconstitution, vaccination, posttransplant.
Referencias bibliográficas
1. Guillaume T, Rubinstein DB, Symann M. Immune reconstitution and immunotherapy after autologous hematopoietic stem cell transplantation. Blood 1998;92:1471-90.
2. Barrett AJ, Rezvani K, Solomon S. New developments in allotransplant immunology. Hematology (Am Soc Hematol Educ Program) 2003:350-71.
3. Sugita K, Soiffer RJ, Murray C, Schlossman SF, Ritz J, Morimoto C. The phenotype and reconstitution of immunoregulatory T cell subsets after T cell depleted allogeneic and autologous bone marrow transplantation. Transplantation 1994;57:1465-73.
4. Storek J. Immune reconstitution after allogeneic marrow transplantation compared with blood stem cell transplantation. Blood 2001;97:3380-9.
5. Chen BJ, Xiuyu C, Sempowski GD, Domen J, Chao NJ . Hematopoietic stem cell dose correlates with the speed of immune reconstitution after stem cell transplantation. Blood 2004;103:4344-52.
6. Koehne ZW, Stockschlaeder M, Zander AR. Phenotype of lymphocyte subsets after autologous peripheral blood stem cell transplantation. Bone Marrow Transplant 1997;19:149-56.
7. Sugita K, Nojima Y, Tachibana K. Prolonged impairment of very late activating antigen-mediated T cell proliferation via the CD3 pathway after T cell depleted allogeneic bone marrow transplantation. J Clin Oncol 1994;94:481-8.
8. Nolte A, Buhmann R, Straka C, Emmerich B, Hallek M. Assessment and characterization of the cytolytic T lymphocyte response against Epstein- Barr virus in patients with non-Hodgkin's lymphoma after autologous peripheral blood stem cell or bone marrow transplants. Bone Marrow Transplant 1998;21:909-16.
9. Roux E, Dumont-Girard F, Starobinski M. Recovery of immune reactivity after T-cell-depleted bone marrow transplantation depends on thymic activity. Blood 2000;96:2299-303.
10. Storek J . Immunity of patients surviving 20 to 30 years after allogeneic or syngeneic bone marrow transplantation Blood 2001;98:3505-12.
11. Small TN, Keever CA, Weiner-Fedus S, Heller G, O'Reilly RJ, Flomenberg N. B cell differentiation following autologous, conventional, or T cell depleted bone marrow transplantation: A recapitulation of normal B cell ontogeny. Blood 1990;76:1647-56.
12. Brenner MK, Wimperis JZ, Reittie JE. Recovery of immunoglobulin isotypes following T cell depleted allogeneic bone marrow transplantation. Br J Haematol 1986;64:125-32.
13. Bolger GB, Sullivan KM, Spencer AM. Myasthenia gravis after allogeneic bone marrow transplantation: Relationship to chronic graft-versus-host disease. Neurology 1986;36:1087-91.
14. Aldouri M, Ruggier R, Epstein O, Prentice HG. Adoptive transfer of hyperthyroidism and autoimmune thyroiditis following allogeneic bone marrow transplantation for chronic myeloid leukaemia. Br J Haematol 1990;74:118-20.
15. Thomas ED, Blume K, Forman SJ. Bacterial infections in hematopoietic cell transplantation. Blackwell Science 1999:537-54.
16. Halverson DC, Schwartz GN, Carter C, Gress RE, Fowler DH. In vitro generation of allospecific human CD8+ T cells of Tc1 and Tc2 phenotype. Blood 1997;90:2089-96.
17. Marsh SG, Parham P, Dupont B. Killer-cell immunoglobulin-like receptor (KIR) Nomenclature report, 2002. Hum Immunol 2003;64:648-54.
18. Childs R, Clave E, Contentin N. Engraftment kinetics after nonmyeloablative allogeneic peripheral blood stem cell transplantation: full donor T-cell chimerism precedes alloimmune responses. Blood 1999;94:3234-41.
19. Ruggeri L, Capanni M, Urbani E. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002;295:2097-100.
20. Ringden O, Barrett AJ, Zhang ML. Decreased treatment failure in recipients of HLA-identical bone marrow or peripheral blood stem cell transplants with high CD34 cell doses. Br J Haematol 2003;121:874-85.
21. Billingham T. The biology of graft-versus-host reactions. En: The Harvey Lectures. New York : Academic Press; 1966. p. 21-78.
22. Korngold R, Friedman TM. Murine models for graft-vs.-host disease. En: Blume KG, Forman SJ, Appelbaum FR, eds. Thomas' Hematopoietic Cell Transplantation. 3 ed. Malden , MA : Blackwell Publishing; 2004.
23. Ferrara JLM, Levy R, Chao NJ . Pathophysiologic mechanism of acute graft-vs.-host disease. Bone Marrow Transplant 1999;25:347-56.
24. Stephanie JL. New approaches for preventing and treating chronic graft-versus-host disease. Blood 2005;11:4200-6.
25. Lorenz E, Schwartz D, Paul A. Association of TLR4 mutations and the risk for acute GVHD after HLA-matched-sibling hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2001;7:384-7.
26. Farag S, Fehniger L. Natural killer cell receptors: new biology and insights into the graft-versus-leukemia effect. Blood 2002;10:1935-47.
27. Molldrem J, Komanduri K. Over expressed differentiation antigens as targets of graft-versus-leukemia reactions. Curr Opin Hematol 2002;9:503-8.
28. Jiang YZ, Barrett J. The allogeneic CD4+ T-cell-mediated graft-versus-leukemia effect. Leuk Lymphoma 1997;28:33-42.
29. Jones SC, Murphy GF, Friedman TM, Korngold R. Importance of minor histocompatibility antigen expression by nonhematopoietic tissues in a CD4+ T cell-mediated graft-versus-host disease model. J Clin Invest 2003;112:1880-6.
30. Clark RE, Dodi IA , Hill SC. Direct evidence that leukemic cells present HLA-associated immunogenic peptides derived from the BCR-ABL b3a2 fusion protein. Blood 2001;98:2887-93.
31. Molldrem S, Dermime KP. Targeted T-cell therapy for human leukemia: Cytotoxic T lymphocytes specific for a peptide derived from proteinase 3 preferentially lyse human myeloid leukemia cells. Blood 1996;88:2450-7.
32. Bellantuono L, Gao SP. Two distinct HLA-A0201-presented epitopes of the Wilms tumor antigen 1 can function as targets for leukemia-reactive CTL. Blood 2002;100:3835-7.
33. Jeffrey A, Paul S. The graft -VS leukemia effect. Implications for post-marrow transplant antileukemia treatment. Am J Pediat Hematol Oncol 1993;15:185-95.
34. Miklos DB, Kim HT, Miller KH. Antibody responses to H-Y minor histocompatibility antigens correlate with chronic graft versus host disease and disease remission. Blood 2005;105:2973-8.
35. Khouri RM, Saliba SA. Giralt. nonablative allogeneic hematopoietic transplantation as adoptive immunotherapy for indolent lymphoma: Low incidence of toxicity, acute graft-versus-host disease, and treatment-related mortality. Blood 2001;98:3595-9.
36. Ferrara JLM, Levy R, Chao NJ. Pathophysiologic mechanisms of acute graft-vs-host disease. Biol Blood Marrow Transplant 1999;5:347-56.
37. Andre-Schmutz I, Le Deist F, Hacein-Bey-Abina S. Immune reconstitution without graft-versus-host disease after hematopoietic stem cell transplantation: A phase I/II study. Lancet 2002;360:130-7.
38. Fowler DH, Breglio J, Nagel G. Allospecific CD4+, Th1/Th2 and CD8+, Tc1/Tc2 populations in murine GVL: Type I cells generate GVL and type II cells abrogate GVL. Biol Blood Marrow Transplant 1996;2:118-25.
39. Bonini C, Ferrari G, Verlezetti S. HSV Tk gene transfer into donor lymphocytes for control of allogeneic graft-versus leukemia. Science 1997;276:1719-24.
40. Pirofski LA, Casadevall A. Use of licensed vaccines for active immunization of the immunocompromised host. Clin Microbiol Rev 1998;11:1-26.
41. Ljungman P, Cordonnier C, deBock R. Immunisations after bone marrow transplantation: Results of a European survey and recommendations from the infectious diseases working party of the European group for blood and marrow transplantation. Bone Marrow Transplant 1995;15:455-60.
42. Centers for Disease Control and Prevention: Guidelines for preventing opportunistic infections among hematopoietic stem cell transplant recipients: Recommendations of CDC, the Infectious Disease Society of America, and the American Society of Blood and Marrow Transplantation. MMWR Morb Mortal Wkly Rep 49(RR-10) 2000:1-125.
43. Versluis DJ, Beyer WE, Masurel N, Wenting GJ, and Weimar W. Impairment of the immune response to influenza vaccination in renal transplant recipients by cyclosporine, but not azathioprine. Transplantation 1986;42:376-9.
44. Hammarstrom V, Pauksen K, Bjorkstrand B, Simonsson B,Oberg G, Ljungman P. Tetanus immunity in autologous bone marrow and blood stem cell transplant recipients. Bone Marrow Transplant 1998;22:67-71.
45. Lin R, Tarr PE, Jones TC. Present status of the use of cytokines as adjuvants with vaccines to protect against infectious diseases. Clin Infect Dis 1995;21:1439-49.
46. Heath AW, Playfair JHL. Cytokines as immunological adjuvants. Vaccine 1992;10:427-34.
47. Santiago -Schwartz F, Divaris N, Kay C, Carsons SE. Mechanisms of tumor necrosis factor-granulocyte macrophage colony stimulating factor induced dendritic cell development. Blood 1993;82:3019-28.
48. Lum LG, Noges JE, Beatty P. Transfer of specific immunity in marrow recipients given HLA-mismatched, T cell-depleted, or HLA-identical marrow grafts. Bone Marrow Transplant 1988;3:399-406.
49. Kato S, Yabe H, Yabe M. Studies on transfer of varicella-zoster-virus specific T-cell immunity from bone marrow donor to recipient. Blood 1990;75:806-9. 50. Centers for Disease Control and Prevention. Guidelines for preventing opportunistic infections among hematopoietic stem cell transplant recipients: Recommendations of CDC, the Infectious Disease Society of America, and the American Society of Blood and Marrow Transplantation. Transplantation 2000;6:705-6.
Recibido: 15 de septiembre del 2006. Aprobado: 6 de octubre del 2006.
Dr. Juan Carlos Jaime Fagundo. Instituto de Hematología e Inmunología. Apartado Postal 8070, Ciudad de La Habana, CP 10800, Cuba. Tel. (537) 6438268, 6438695, 6434214, Fax (537) 442334. e-mail: ihidir@hemato.sld.cu
1Instituto de Hematología e Inmunología.
2Hospital Pediátrico Docente "William Soler".

