










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter v.24 n.1 Ciudad de la Habana ene.-abr. 2008
PRESENTACION DE CASOS
Uso de la prueba de rupturas cromosómicasen el estudio de la anemia de Fanconi
Use of the chromosome rupture test in the study of Fanconi's anemia. Preliminary results
Dra. Kalia Lavaut Sánchez; Dr. Juan C. Jaime Facundo; Dr. Aramís Núñez Quintana; Dra. Valia Pavón Morán; Prof. Porfirio Hernández Ramírez; Dr. Sergio Machín
Instituto de Hematología e Inmunología. Cuba. E-mail:ihidir@hemato.sld.cu
RESUMEN
La anemia de Fanconi es un desorden genético recesivo con ambos patrones de herencia, autosómico y ligado al sexo, caracterizada por diferentes malformaciones congénitas, fallo de médula ósea y una elevada predisposición a desarrollar tumores sólidos y leucemia mieloide aguda. Es una enfermedad monogénica con expresión citogenética dada por inestabilidad cromosómica tanto espontánea como provocada por agentes inductores de enlaces cruzados en las cadenas de ADN. Se presentan 2 pacientes masculinos, hermanos, de 5 y 7 años de edad, con malformaciones congénitas e insuficiencia medular. Se les realizó el estudio de rupturas cromosómicas con el uso del diepoxibutano y se observaron múltiples rupturas y figuras radiales, lo que confirmó el diagnóstico.
Palabras clave: anemia de Fanconi, rupturas cromosómicas, diepoxibutano.
ABSTRACT
Fanconi's anemia is a recurrent genetic disorder with both patterns of heredity, autosomal and linked to sex. It is characterized by different congenital malformations, bone marrow failure and an elevated predisposition to develop solid tumors and acute myeloid leukemia. It is a monogenic disease with cytogenetic expression given by chromosomal instability, both spontaneous and provoked by agents inducing cross-links in the DNA chains. Two male siblings aged 5 and 7 years old, with congenital malformations and medullar insufficiency were presented. The study of chromosome rupture was conducted by using diepoxybutane. Multiple ruptures and radial figures were observed, which confirmed the diagnosis.
Key words: Fanconi's anemia, chromosome ruptures, diepoxybutane.
INTRODUCCIÓN
La anemia de Fanconi (AF) es un síndrome de fragilidad cromosómica, autosómico recesivo y ligado al X, que presenta malformaciones congénitas muy diversas y en diferentes órganos en el 70 % de los casos, insuficiencia medular progresiva y tendencia a enfermedades malignas sobre todo leucemia mieloide (LMA) y tumores sólidos.
Esta entidad fue descrita en 1927 por el pediatra suizo Guido Fanconi, en 3 hermanos con varias malformaciones congénitas asociadas con astenia, infecciones a repetición y sangramientos espontáneos por fallo en la función de la médula ósea.1
Es la anemia aplásica más frecuente en la infancia y afecta a 1:360.000 nacimientos. La proporción de varones y hembras es de 3:1. 2 El 75 % de los casos se diagnostica entre los 4 y 14 años, aunque se han reportado desde el nacimiento hasta los 48 años, con una edad media de 8 años.
Entre las características clínicas se señala la baja talla de comienzo prenatal (60 %), pigmentación carmelita de la piel (65 %), microcefalia (30 %), ptosis palpebral (25 %), estrabismo, nistagmo y microftalmía, defecto radial (50 %) que incluye hipoplasia, aplasia del pulgar, pulgar supernumerario, aplasia o hipoplasia de radio, anomalías de los tractos urinario y renal (25 %) con hipoplasia acompañada o no de malformaciones renales, uréteres dobles, hipospadias, micropene, testículos pequeños, criptorquídia y pancitopenia de grado variable que puede llegar a la aplasia medular.3
Genéticamente, la AF es una enfermedad muy heterogénea. Hasta el momento, se han descrito 12 grupos de complementación (A, B, C, D1, D2, E, F, G, I, J, L y M), definidos por estudios de fusión celular, de los cuales 11 de ellos han sido identificados. De estos, el grupo de complementación FANCA es el más frecuente y presenta un amplio espectro de mutaciones, siendo las grandes deleciones una de las principales.4 La mayoría de las proteínas de la AF (A, B, C, E, F, G, I, L, M) forman un complejo nuclear necesario para la posterior activación del D2 mediante la ubiquitinación de la lisina 561 en respuesta a los agentes de enlaces cruzados. El D2 activado unido con otras proteínas tiene un importante papel en la reparación del ADN.5
Otros rasgos celulares son la sensibilidad frente al daño oxidativo, fallos en el ciclo celular, niveles elevados de apoptosis, regulación deficiente de la transcripción y disfunción de los telómeros, estructuras que se encuentran situadas en el extremo de los cromosomas, cuya función es protegerlo de la degradación, impedir que se unan entre sí y favorecer la diferenciación correcta de los mismos durante los procesos de división celular. En estos pacientes existe un acortamiento de los telómeros, que es el responsable de la inestabilidad del gen, la apoptosis celular, las alteraciones hematológicas y el cáncer, lo cual se ha demostrado por técnicas cuantitativas de hibridación in situ fluorescente (FISH).6-8
La confirmación diagnóstica de la enfermedad se realiza mediante estudios de fragilidad cromosómica (roturas de cromosomas) inducidas por agentes clastógenos como el diepoxibutano (DEB), que generan enlaces cruzados en las cadenas de ADN. Para realizar este estudio, los linfocitos T de la sangre se ponen en cultivo en presencia y ausencia de DEB, exponiéndose posteriormente a colchicina, siguiendo los métodos citogenéticos convencionales. En este prueba se tiene en cuenta el número, tipo de roturas cromosómicas detectadas en cada célula y la distribución de estas, efectuándose el cálculo del porcentaje de células con roturas, el número medio de roturas por célula y presencia de figuras radiales, lo que permite confirmar el diagnóstico y detectar mosaicismos, es decir, la presencia de 2 subpoblaciones celulares en los cultivos tratados con DEB, una sin roturas cromosómicas y otra con muchas roturas por célula. Este estudio no es útil para detectar el estado de portador.9,10
El objetivo de este trabajo es presentar dos hermanos con anemia de Fanconi, su cuadro clínico y mostrar los resultados obtenidos con el uso de la técnica con diepoxibutano, estandarizada en nuestro laboratorio.
Presentación de casos
Caso No 1:
Paciente de 7 años de edad, masculino, que asistió a consulta a los 6 años de edad por anemia, con antecedentes prenatales y perinatales negativos.
El examen físico mostró baja talla (de comienzo prenatal), microcefalia, microftalmía, coloración bronceada de la piel, manchas café con leche en el tórax, e hipoplasia de ambos pulgares.
Los exámenes hematológicos evidenciaron: hemoglobina 84 g/L, reticulocitos 0,8 %, plaquetas 60 x 109 y leucocitos 5,4 x 109. En los estudios evolutivos siempre se confirmó pancitopenia.
En el medulograma se encuentra una celularidad disminuida con ausencia de megacariocitos, pero con el resto de los sistemas normales.
En el estudio citogenético en sangre periférica con el uso de diepoxibutano, para investigar la existencia de rupturas cromosómicas, se analizaron 30 metafases, con fórmula cromosómica de 46, XY y se observó un total de 19 rupturas, imagen de trirradio del cromosoma 2 y una tasa de ruptura de 0,63.
El enfermo requirió transfusiones frecuentes y falleció a los 9 años de edad debido a complicaciones hemorrágicas.
Caso No 2:
Paciente masculino, de 5 años de edad con antecedentes de parto pretérmino, que asistió a consulta por malformaciones congénitas y el antecedente de anemia de Fanconi en su único hermano.
Al examen físico se encontró baja talla de comienzo prenatal, microcefalia, microftalmía, color bronceado de la piel, pulgares de localización proximal en ambas manos, hipospadias y criptorquídia.
Tenía una hemoglobina de 107 g/L, plaquetas 160 x 109, leucocitos 7,8 x 109. Los hemogramas evolutivos mostraron descenso de las cifras de hemoglobina y plaquetas, mientras que los leucocitos se mantenían en valores normales.
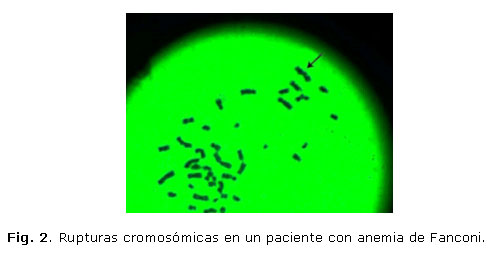
En el estudio citogenético en sangre periférica con el uso de diepoxibutano, se analizaron 30 metafases, con una fórmula cromosómica 46, XY y se observaron 22 rupturas, cromosomas acéntricos, 3 figuras radiales y una tasa de ruptura de 0,73 (figs. 1 y 2).
Evolutivamente el paciente mantiene la hemoglobina y cifras de plaquetas bajas, con tratamiento de esteroides, sin requerimiento transfusionales.
DISCUSIÓN
La anemia de Fanconi es una enfermedad con un patrón de herencia frecuentemente autosómico recesivo, por lo que en esta situación se necesitan las 2 copias del gen mutado para que se produzca la enfermedad. Si 2 individuos portadores de la mutación tienen descendencia, existe la probabilidad de que el 25 % de la misma tengan ambas copias no funcionales; estos son los enfermos de AF. Esta probabilidad se comporta como evento independiente para cada embarazo. La existencia de casos ligados al sexo es una condición extremadamente infrecuente.
Las malformaciones congénitas están presentes en el 60-75 % de los niños.11,12 En nuestros pacientes, las alteraciones del pulgar, hiperpigmentación de la piel, baja talla de comienzo prenatal y la edad de aparición de las manifestaciones hematológicas, coinciden con la literatura revisada.13
El diagnóstico basado en observaciones clínicas puede presentar dificultad debido a la gran variabilidad de síntomas que muestran, unido a que un porcentaje variable de los pacientes con AF no muestran malformación alguna, motivo por el cual los estudios citogenéticos y moleculares resultan de gran importancia.
La confirmación diagnóstica de la enfermedad se realiza mediante estudios de fragilidad cromosómica (prueba de rupturas) inducidas por agentes clastógenos. En nuestro laboratorio se introdujo el estudio con el uso del diepoxibutano en cultivo de 72 horas de linfocitos, que permite confirmar la anemia de Fanconi en ambos pacientes. En una proporción de pacientes, menos del 10 % el estudio puede resultar negativo debido al mosaicismo somático, estos individuos presentan 2 poblaciones de células T en sangre, una de ellas sensible al DEB y otras no; esta población resistente es el resultado de sucesos recombinacionales o de conversión génica, que revierten el alelo patogénico a su estado normal, lo cual dificulta el diagnóstico. En aquellos pacientes con alta sospecha clínica de AF y negatividad en el estudio, debe repetirse el estudio en cultivo de fibroblastos para evidenciar las rupturas cromosómicas.
La introducción de esta técnica nos permite realizar diagnóstico de la enfermedad aún sin estar presentes las alteraciones hematológicas.
Como complemento del diagnóstico citogenético en la AF, resulta conveniente determinar la mutación responsable de la enfermedad. Mediante el análisis mutacional se pueden identificar los portadores de la enfermedad y realizar estudios de diagnóstico prenatal o preimplantacional. Todas estas pruebas permiten confirmar el diagnóstico de la enfermedad, aspecto muy importante para tomar la conducta terapéutica.
La terapia de elección para los pacientes con AF se basa en el trasplante de progenitores hematopoyéticos obtenidos a partir de donantes histocompatibles sanos. Los mejores resultados se obtienen cuando el trasplante se realiza en edades tempranas y cuando el paciente ha recibido un número reducido de transfusiones.
REFERENCIAS BIBLIOGRÁFICAS
1. Fanconi G. Familiare infantile perniziosaartige anemia. Jahrbunch Kinder 1927;1117:257-61.
2. Nelson WE, Berhman RE, Liegman RM, Arvin AM. Tratado de Pediatría. 15 ed. Madrid: McGraw Hill Interamericana; 2000.
3. Kenneth Lyons J. S mith's recognizable patterns of human malformation. 6 ed. Philadelphia: Saunders; 2006 pp. 362-3.
4. Hamosh A, Scott AF, Amberger J, Bocchini C, Valle D, McKusick VA. Online Mendelian Inheterance in Man, (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res 2002;30:52-5
5. Pasquini R. Fanconi anemia. Arch Med Int Uruguay 2007;29(Supl1):557-9.
6. Hanson H, Mathew CG, Docherty Z, Mackie Ogilvie C. Telomere shortening in Fanconi anemia demonstrated by a direct FISH approach. Cytogenet Cell Genet 2001;93:203-6.
7. Callén E, Samper E, Ramírez MJ, Creus A, Marcos R, Ortega JJ. Breaks at telomeres and TRF2-independent end fusions in Fanconi anemia. Hum Mol Genet 2002;11:439-44.
8. Callén E. Biología y genética molecular de la anemia de Fanconi. [Tesis]. Barcelona: Universidad Autónoma de Barcelona; 2004. Disponible en: http://www.tdx.cesca.es/TDX-0726105 -234012/.
9. Callén E, Surrallés J. ¿Cómo se diagnostica la enfermedad? Boletín Informativo para especialistas y familiares de pacientes. Grupo Español para el estudio y tratamiento de la anemia de Fanconi 2002;1:4-6.
10. Red Nacional para la anemia de Fanconi. Guía básica para el diagnóstico y seguimiento de pacientes con anemia de Fanconi. [Citado el 1 de Julio del 2007]. Disponible en: http://www.redfanconi.net.
11. D´Andrea A, Dahl N, Guinan E, Shimaruma A. Marrow failure. Hematology 2002;12:58-72.
12. Sagaseta de Ilurdoz M, Molina J, Lezáun I, Valiente A, Durán G. Anemia de Fanconi. Consideraciones actuales. Anales Sis San Navarra 2003;26(1): [Citado el 10 enero 2008]. Disponible en: <http://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S1137 -66272003000100006&lng=es&nrm=iso>.
13. Butturini A, Gale RP, Verlander PC, Alder-Brecher B, Gillio AP, Auerbach AD. Hematologic abnormalities in Fanconi anemia: an International Fanconi Anemia Registry Study. Blood 1994;84:1650-5.
Recibido: 15 de diciembre del 2007.
Aprobado: 3 de enero del 2008.
Dra. Kalia Lavaut Sánchez.
Instituto de Hematología e Inmunología. Apartado Postal 8070, Ciudad de La Habana, CP 10800, Cuba. Tel (537) 6438268, 6438695. Fax (537) 6442334. e-mail: ihidir@hemato.sld.cu


{kind=link}