










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter v.25 n.2 Ciudad de la Habana Mayo-ago. 2009
ARTÍCULO DE REVISIÓN
Moléculas de adhesión. Importancia en el síndrome coronario agudo
Significance of adhesion molecules in acute coronary syndrome
DraC. Consuelo Macías Abraham; Lic. Lázaro O. del Valle Pérez; DrC. Prof. Porfirio Hernández Ramírez; DrC. Prof. José M. Ballester Santovenia
Instituto de Hematología e Inmunología. Ciudad de La Habana, Cuba.
RESUMEN
Se presenta una revisión de los conceptos actuales de la ateroesclerosis como fenómeno inflamatorio crónico del endotelio vascular y de la importancia de la participación de las moléculas de adhesión endoteliales en las formas clínicas principales del síndrome coronario agudo: la angina inestable y el infarto agudo del miocardio. Se describen los resultados obtenidos por diferentes autores en el estudio de las moléculas de adhesión en este síndrome, su importancia en el diagnóstico asociado con los parámetros séricos convencionales y su potencial terapéutico.
Palabras clave: moléculas de adhesión, síndrome coronario agudo, angina inestable, infarto agudo del miocardio.
ABSTRACT
Present paper is a review of current concepts of atherosclerosis as a chronic inflammatory phenomenon of vascular endothelium and of significance involvement of endothelial adhesion cells in the main clinical ways of acute coronary syndrome: unstable angina and myocardial acute infarction. Different authors describe the results achieved in adhesion molecules study in this syndrome, its significance in diagnosis associated with conventional serum parameters and its therapeutical potential.
Key words: Adhesion molecules, acute coronary syndrome, unstable angina, myocardial acute infarction.
INTRODUCCIÓN
La cardiopatía isquémica es un trastorno del miocardio provocado por el desequilibrio entre los requerimientos del músculo cardíaco y el flujo sanguíneo coronario, cuya causa fundamental es la aterosclerosis. 1
En décadas anteriores, el endotelio vascular se consideraba como un revestimiento no adhesivo de los vasos sanguíneos, cuya función consistía en prevenir la coagulación sanguínea y separar el espacio vascular de los tejidos. En comparación con otros tipos celulares, se pensaba que las células endoteliales eran menos activas, complejas e interesantes. En la actualidad, está demostrado que el endotelio vascular participa activamente en una amplia variedad de procesos fisiológicos como la inflamación y la respuesta inmunológica. 2-5
Con las profundas y sólidas transformaciones económicas y sociales llevadas a cabo en Cuba, se han logrado desplazar de las causas de mortalidad las enfermedades típicas del subdesarrollo.6,7 Actualmente, las enfermedades crónicas no transmisibles ocupan los primeros lugares de morbimortalidad y dentro de ellas, las enfermedades cardiovasculares como la cardiopatía isquémica y el síndrome coronario agudo (SCA). 8
Las 2 hipótesis clásicas sobre el origen de la aterosclerosis por incrustación de material trombolítico y por imbibición de material lipídico, se han integrado en una hipótesis multifactorial más compleja que explica el proceso ateromatoso. Esta hipótesis se basa en la llamada respuesta de la pared a la lesión o respuesta de reparación celular, que según la misma no son solo la trombosis o los lípidos los factores primordiales en el proceso ateromatoso, sino también las proteínas sanguíneas, otros tipos celulares y la secuencia de lesiones que se desarrollan en la propia pared vascular. 9
La lesión endotelial producida por la aterosclerosis es un factor esencial en la evolución, progresión y manifestación clínica del SCA. Existen diferentes grados de lesión endotelial, desde una alteración meramente funcional sin cambios morfológicos significativos, hasta lesiones más profundas, 10,11 que provocan una serie de eventos funcionales con participación activa de las moléculas de adhesión, que pueden desencadenar isquemia.
La aterosclerosis es una enfermedad del sistema vascular arterial que ha sido definida por la OMS como una combinación variable de cambios de la íntima que provocan la acumulación focal de lípidos, carbohidratos complejos, sangre y sus productos, tejidos fibrosos y depósito de calcio asociado con cambios en la media.12 Se caracteriza por un desarrollo lento, progresivo y silencioso hasta que la lesión alcanza un tamaño capaz de producir isquemia, lo que afecta a las arterias de grande y mediano calibre (aorta, coronarias, carótidas, ilíacas y femorales) y a nivel cardiovascular es la causa principal de la cardiopatía isquémica. 1
DESARROLLO DE LA ATEROSCLEROSIS
En la pasada década han existido enormes progresos en la comprensión de la naturaleza de la aterosclerosis. El desarrollo de una placa aterosclerótica es un proceso complejo, que comienza con la disfunción endotelial asociada con otros factores como la hipercolesterolemia, el tabaquismo, la hipertensión, la hiperhomocisteinemia y los trastornos del metabolismo de la glucosa. Esta disfunción incluye el incremento de la permeabilidad a las lipoproteínas y otros constituyentes del plasma, la cual es mediada por el óxido nítrico, el factor de diferenciación y crecimiento plaquetario (PDGF, del inglés, platelet diferentiation growth factor), de la prostaciclina, la angiotensina II y la endotelina; la sobreexpresión de las moléculas de adhesión VCAM-1, ICAM-1 y selectinas y la migración de los leucocitos en el espacio subendotelial mediado por lipoproteínas de baja densidad (LDL, del inglés, low density lipoproteins) oxidadas y citocinas como el PDGF, el MCP-1 y el factor estimulador de colonias de macrófagos (MCSF, del inglés, macrophages colony stimulator factor). Esto es seguido de la migración de las células del músculo liso (estimuladas por el PDGF y el TGFß), la activación de las célulasT (mediada por TNFa y la IL-2), la formación de células espumosas (mediado por LDL oxidadas, MCSF, TNF e IL-1) y la agregación y adherencia plaquetaria (estimulada por el tromboxano A2 y factores tisulares). Las células del músculo liso forman una capa fibrosa que confiere estabilidad mecánica a la placa y separa el núcleo trombogénico rico en lípidos de la luz vascular y la sangre circulante. La placa puede permanecer intacta y estable o romperse, lo que da lugar a la trombosis y causa un SCA, infarto agudo del miocardio (IAM) o angina inestable (AI), dependiendo de diferentes factores, entre los cuales, el más importante es su composición. El tamaño de la placa es de menor importancia en el riesgo de padecer un SCA; la ruptura de la capa fibrosa ocurre debido a un adelgazamiento de esta causada por un influjo de macrófagos activados que liberan metaloproteinasas y otras enzimas proteolíticas, estimulados por células inflamatorias, especialmente los linfocitos T. Estas enzimas causan degradación del tejido fibroso de la capa, lo cual puede resultar en la formación de trombos y oclusión de la arteria. Las placas estables tienen una capa fibrosa compacta, un pequeño núcleo lipídico y escasas células inflamatorias. Sin embargo, las placas vulnerables tienen un alto contenido lipídico, numerosas células inflamatorias y una delgada capa fibrosa con poca cantidad de colágeno y células del músculo liso. 13
En los últimos años se ha demostrado que en las placas ateromatosas humanas existen células inflamatorias como los linfocitos T, los monocitos y los macrófagos; proteínas inflamatorias como citocinas y quimocinas; y moléculas de adhesión endoteliales como signo de respuesta inflamatoria de las células vasculares, inmunoglobulinas y factores activadores del complemento, lo que sugiere la participación de los mecanismos inmunológicos en su formación. Por todo lo anterior, se han estudiado las lesiones ateromatosas como una forma de respuesta fibro-inflamatoria. 10,11,13-16
MARCADORES SÉRICOS INFLAMATORIOS EN EL SCA
En la clínica, se ha demostrado la utilidad de la detección sérica de diferentes marcadores de la inflamación en el SCA, como la proteína C reactiva (predictiva de complicaciones tardías como la restenosis, el reinfarto y la muerte por eventos cardíacos que permite aplicar terapia antiinflamatoria con ácido acetil salicílico y estatinas), la amiloide A, la troponina T (correlacionada con un incremento de complicaciones tempranas que permite que el paciente se beneficie de una terapia agresiva con intervención coronaria aguda y antagonistas del receptor GpIIb/IIIa);16 y citocinas como las IL-1 y 6, que se encuentran asociadas con la patogénesis del SCA y su diagnóstico diferencial, por lo que algunos de ellos se han utilizado como marcadores diagnósticos y pronósticos.17-23 En este síndrome también se han evaluado los niveles de las moléculas de adhesión endoteliales solubles VCAM-1, ICAM-1 y E-selectina, como marcadores séricos característicos de la disfunción endotelial e inflamación, con resultados controvertidos. 24-28 Se ha descrito la concentración elevada de CD40L soluble en el SCA, el que se correlacionó positivamente con las concentraciones de ICAM-1; y negativamente con las concentraciones de colesterol y lipropoteínas de alta densidad. 29
Otros autores han planteado que la respuesta inflamatoria aguda en pacientes con dolor precordial, caracterizada por elevadas concentraciones de ICAM-1 soluble, IL-6 y proteína C reactiva, desencadena en poco tiempo un SCA, y que en el seguimiento de enfermos con SCA, la molécula ICAM-1 mostró valores elevados que se mantuvieron hasta 3 meses, por lo que este marcador pudiera ser un importante factor de riesgo en el desarrollo del SCA. 30
También se ha descrito el incremento de la concentración de la molécula PECAM-1 en la angina estable, la angina inestable e IAM, en relación con los sujetos sanos, pero no existieron diferencias entre los 3 síndromes coronarios, por lo que pudiera ser un marcador en el diagnóstico precoz. 31
Nuestros resultados han mostrado niveles elevados de ICAM-1 y VCAM-1 solubles en pacientes con IAM, a la admisión y 10 días después. Se observó un patrón característico de liberación de estas moléculas de adhesión: inicialmente estas moléculas se elevaron en la fase aguda del daño isquémico miocárdico, momento en que el flujo sanguíneo coronario está disminuido. Sin embargo, el patrón de E-selectina durante el seguimiento longitudinal difirió del patrón anteriormente descrito para las moléculas ICAM-1 y VCAM-1, ya que no mostró diferencias significativas comparado con los valores normales al momento de la admisión y 10 días después, a pesar de la persistencia de los valores incrementados de ICAM-1 y VCAM-1. En los pacientes con AIN se obtuvieron niveles elevados de VCAM-1, ICAM-1 y E-selectina, a la admisión y 10 días después, pero los valores obtenidos de ICAM-1 no mostraron diferencias significativas comparados con los controles normales y pacientes con IAM. 32
Niveles elevados de ICAM-1 han sido previamente reportados en el IAM 24,25,33 y en la AIN. 26,34-36 En un estudio reciente se comunicó que pacientes con IAM mostraron niveles elevados de ICAM-1 y E-selectina en la circulación periférica, pero los niveles de E-selectina alcanzaron sus valores máximos entre las 4 y 6 horas posteriores a la admisión y retornaron a sus niveles basales a las 24 horas.24 Los estudios in vitro también han demostrado diferencias en la cinética de la expresión endotelial de la E-selectina e ICAM-1. 37 Sin embargo, otras investigaciones no han evidenciado incremento de la E-selectina en el IAM. 25,26
La activación de la microcirculación es el principal componente de la respuesta inflamatoria. El endotelio vascular desarrolla y expresa moléculas que inician la inmigración local de leucocitos.38,39 La molécula ICAM-1 puede ser liberada (específica e inespecíficamente) por el tejido dañado o inflamado como consecuencia de una proteólisis inespecífica.40 Esta observación podría explicar los niveles elevados de ICAM-1 en pacientes con síndrome coronario agudo y sus valores más elevados en el IAM. 32
La VCAM-1 es expresada por el endotelio arterial en las lesiones ateroescleróticas tempranas en un modelo experimental de aterosclerosis en conejo (conejo Watanabe) y podría ser responsable de la atracción de las células mononucleares que desarrollan la lesión ateroesclerótica.21 Otros autores han mostrado previamente que la VCAM-1 es expresada ampliamente sobre células endoteliales de arterias ocluidas durante la ateroesclerosis acelerada.41 La ateroesclerosis coronaria es la causa más frecuente de enfermedad cardíaca por isquemia y la ruptura de las placas por trombosis es la principal causa de los síndromes coronarios agudos como la AIN, IAM y muerte súbita.42 Estas observaciones podrían explicar la elevada concentración de VCAM-1 en los síndromes coronarios agudos y por qué existe una significativa disminución en las concentraciones séricas de esta molécula 10 días después en la AIN cuando el flujo sanguíneo coronario ha mejorado.32
Nuestros resultados han demostrado un significativo incremento de la concentración de E-selectina en el grupo de pacientes con AIN a la admisión y 10 días después.32 La E-selectina es particularmente interesante debido a que esta molécula se encuentra solamente en el endotelio activado, en contraste con otras moléculas de adhesión que tienen una más amplia distribución tisular. Por lo tanto, la demostración de E-selectina soluble en la sangre se puede considerar como una evidencia concluyente de activación endotelial. Esta molécula de adhesión es biológicamente activa mediando la adhesión de neutrófilos a la superficie endotelial, así como la de monocitos, eosinófilos, basófilos y células NK.43,44 La E-selectina facilita la fase temprana de adhesión de los polimorfonucleares a la célula endotelial, lo que constituye un marcador sérico temprano de la respuesta inflamatoria promoviendo el daño celular por isquemia.45,46 Episodios de isquemia breve durante la angioplastia coronaria permiten el estímulo soluble capaz de inducir la expresión de integrinas en el neutrófilo 47,48 y también se ha observado en estudios realizados en pacientes con AIN, que la aparición del dolor torácico después de las 48 horas de la angiografía coronaria, está relacionado con valores significativamente más altos de activación del neutrófilo, lo que sugiere que el grado de activación está relacionado con próximos episodios de angina en reposo.47
Por otra parte, las concentraciones de interleucina-1 beta (IL-1ß) se han encontrado elevadas en los pacientes con enfermedad cardíaca isquémica, en particular en aquellos con enfermedad arterial coronaria mínima y angina.49 La función precisa de la IL-1ß en la enfermedad arterial coronaria permanece indeterminada, pero durante la respuesta inflamatoria, las células endoteliales expresan E-selectina en respuesta a la IL-1.49,50 Teniendo en cuenta estas evidencias, los episodios isquémicos podrían actuar como desencadenantes de un mecanismo secuencial representado por la activación endotelial con expresión de E-selectina y su liberación a la sangre, niveles incrementados de IL-1ß que promueven la expresión de E-selectina por las células endoteliales y activación de los neutrófilos por la E-selectina soluble, lo que provoca daño celular isquémico y consecuentemente, más liberación de E-selectina por la célula endotelial activada (figura).32 Probablemente estas reacciones integradas podrían explicar por qué la E-selectina se comporta de una manera diferente a la reportada en el IAM por algunos autores. 24,32
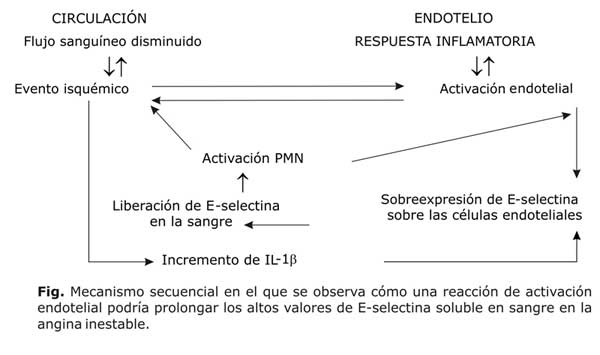
Diferentes hallazgos de otros autores facilitan la comprensión de nuestros resultados si consideramos que el IAM tiene asociada una respuesta inflamatoria mayor que la AIN 23,25,26,51-53 y que la AIN se asocia con un mecanismo inflamatorio no relacionado con la presencia de necrosis ni con la extensión de las lesiones coronarias. 17
RELACIÓN CON EL DAÑO MIOCÁRDICO
En nuestro estudio no se pudo demostrar relación entre los niveles incrementados de las moléculas de adhesión y los resultados de las enzimas cardíacas y los cambios electrocardiográficos.32 Esto sugiere que los altos niveles de las moléculas de adhesión no están asociados con la severidad del daño miocárdico medido, al menos por estos parámetros, en el síndrome coronario agudo, lo que concuerda con lo señalado por otros autores para otros marcadores séricos relacionados con su diagnóstico y pronóstico,17,53 y con la evidencia de que elevaciones específicas de los niveles de E-selectina podrían indicar activación o daño del endotelio como componente particular de una patología -una enfermedad, un trastorno- determinada. 32,45
La oclusión aguda de una arteria coronaria por la generación de un trombo lleva implícita la activación plaquetaria, su hiperreactividad, la adhesión local a la pared del vaso y la formación de agregados por la unión del fibrinógeno soluble con la molécula de adhesión GPIIb/IIIa en la superficie plaquetaria. Estudios con AcMo específicos han demostrado que las plaquetas se adhieren a los monocitos por la vía de la interacción P-selectina/ PSGL-1, receptores de las plaquetas y monocitos respectivamente, por lo que las plaquetas activadas son un poderoso sustrato adhesivo para el reclutamiento de los monocitos en el endotelio vascular. 31
POTENCIAL TERAPÉUTICO
Un excelente AcMo anti gpIIbIIIa plaquetaria ha sido usado para prevenir restenosis después de la angioplastia y un gran número de potentes péptidos de bajo peso molecular y antagonistas no peptídicos se encuentran en desarrollo. Se ha seleccionado un gran número de moléculas para intervenciones terapéuticas como son: la P y E selectinas y otras integrinas como LFA-1, Mac-1, avß3, VLA-4 y a4ß7, L-selectina, CD44 y otras implicadas en los mecanismos inmunes y de la respuesta inflamatoria.54
Ligandos solubles sintéticos de selectinas e integrinas se utilizan para bloquear las funciones adhesivas de estas moléculas e inhibir la inflamación. Las drogas antinflamatorias no esteroideas inhiben la expresión de L-selectina y son muy utilizadas; drogas como el piroxicam y el meloxicam inhiben la translocación de Mac-1 hacia la membrana celular, así como la activación de integrinas inducida por quimocinas. 55
La pentoxifilina inhibe los fenómenos de adhesión celular mediados por integrinas e interfiere con la activación de las ß1 integrinas. La específica inhibición de la síntesis de integrinas y selectinas por oligonucleótidos antisentidos, es otro interesante avance en la terapia antiadhesiva, pero existen problemas a resolver antes de su aplicación práctica. 55
Estas observaciones abren nuevos caminos para una politerapia integrada y racional futura, que permita controlar los principales factores desencadenantes del proceso inflamatorio-oclusivo vascular en las diferentes formas clínicas del SCA.
REFERENCIAS BIBLIOGRÁFICAS
1. Wick G, Schett G, Amberger A, Kleindienst R, Xu Q. Is atherosclerosis an inmunologically mediated disease? Immunol Today 1995;16:17-33.
2. Bevilacqua MP. Endothelial leukocyte adhesion molecules. Ann Rev Immunol 1993;11:767-804.
3. Pober JS, Cotran RS. The role of endothelial cells in inflammation. Transplantation 1990;50:537-44.
4. Zimmerman GA, Prescott SM. Endothelial cells interactions with granulocytes: Tethering and signalling molecules. Immunol Today 1992;3:93-9.
5. Butcher EC. Leukocyte endothelial cells recognition. Three or more steps to specificity and diversity. Cell 1991;67:1033-37.
6. Von Andrian UH, Mackay CR. Advances in Immunology: T cell function and migration. Two sides of the same coin. N Engl J Med 2000;343:1020-34.
7. Verdecia FF. Sociedad y salud. La Habana: Pueblo y Educación; 198. p. 22-84.
8. Cuba. Ministerio de Salud Pública. Dirección Nacional de Estadísticas. Informe Anual. Ciudad de La Habana; 2003.
9. Fuster V, Bediman L. The pathogenesis of coronary artery disease and the acute coronary syndromes. N Engl J Med 1992;326:242-50.
10. Connor W. Prevention complication and treatment in heart disease. Philadelphia: W Saunders; 1980. p. 1221-78.
11. Stein JH. Medicina Interna. La Habana: Ed. Científico-Técnica; 1984. p. 546-52.
12. Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cells subsets. Trends Immunol 2001;22:633-40.
13. Reiner Z, Tedeschi-Reiner E. New information on the pathophysiology of atherosclerosis. Lijec Vjesn 2001;123:26-31.
14. Jerzak P. The role of adhesion molecules in the inmunopathogenesis of atherosclerosis. Pol Tyg Leg 1994;49:357-9.
15. Plutzky J. Inflammatory pathways in atherosclerosis and acute coronary syndromes. Am J Cardiol 2001;18,88:10K-15K.
16. Link A, Bohm M, Nickenig G. Acute coronary syndrome-better risk stratification by determination of inflammatory parameters? Med Klin 2002;97:63-9.
17. Liuzzo G, Biasucci LM, Callimore JR, Grillo RL, Rebuzzi AG, Pepys MB, et al. The prognostic value of C-reactive protein and serum amyloid A protein in severe unstable angina. N Engl J Med 1994;331:417-24.
18. Fuster V, Bediman L. The pathogenesis of coronary artery disease and the acute coronary syndromes. N Engl J Med 1992;326:242-50.
19. Sluiter W. Leukocyte adhesion molecules on the vascular endothelium: Their role in the pathogenesis of cardiovascular disease and the mechanism underlying their expression. J Cardiovascular - Pharmacol 1993;22:537-44.
20. Wang-X. Interleukin-1 beta induces expression of adhesion molecules in human vascular smooth muscle cells and enhances adhesion of leukocytes to smooth muscle cells. Atherosclerosis 1995;115:89-98.
21. Gimbrone MA. Vascular endothelium: An integrator of pathophysiologic stimuli in atherosclerosis. Am J Cardiol 1995;75:678-708.
22. Jerzak P. The role of adhesion molecules in the inmunopathogenesis of atherosclerosis. Pol Tyg Leg 1994;49:357-59.
23. Bazzino O. Valor pronóstico de la proteína C reactiva en la angina inestable. Rev Esp Cardiol 2001;54:1-6.
24. Yi-Heng Li, Jeng-Kai T, Wei-Chun T, Liang-Miin T, Li-Jen L, Jyh-Hong Ch. Elevation of soluble adhesion molecules is associated with the severity of myocardial damage in acute myocardial infarction. Am J Cardiol 1997;80:1218-21.
25. Shyu KG, Chang H, Lin CC, Kuan P. Circulating intercellular adhesion molecule-1 and E-selectin in patients with acute coronary syndrome. Chest 1996;77:543-49.
26. Mulvihill N, Foley JB, Ghaisas N, Murphy R, Crean P, Walsh M. Early temporal expression of soluble cellular adhesion molecules in patients with unstable angina and subendocardial myocardial infarction. Am J Cardiol 1999;83:1265-67.
27. Xie Y, Zhou T, Shen W, Lu G, Yin T, Gong L. Soluble cell adhesion molecules in patients with acute coronary syndrome Chin Med J Engl 2000;113:286-8.
28. Futterman LG, Lemberg L. Novel markers in the acute coronary syndrome: BNP, IL-6, PAPP-A. Am J Crit Care 2002;11:168-72.
29. Peng DQ, Zhao SP, Li YF, Li J, Zhou HN. Elevated soluble CD40 ligand is related to endothelial adhesion molecules in patients with acute coronary syndrome. Clin Chim Acta 2002;319:19-26.
30. O' Malley T, Ludlam CA, Riemermsa RA, Fox KA. Early increase in levels of soluble inter-cellular adhesion molecule-1 (sICAM-1); potential risk factor for the acute coronary syndromes. Eur Heart J 2001;22:1226-34.
31. Soeki T, Tamura Y, Shinohara H, Sakabe K, Onose Y, Fukuda N. Increased soluble platelet/endothelial cell adhesion molecule-1 in the early stages of acute coronary syndromes. Int J Cardiol 2003;90:261-8.
32. Macías C, Villaescusa R, del Valle L, Boffill V, Cordero G, Hernández A, et al. Moléculas de adhesión endoteliales ICAM-1, VCAM-1 y E-selectina en pacientes con síndrome coronario agudo. Rev Esp Cardiol 2003;56:137-44.
33. Wallen NH, Held C, Rehnquist N, Hjemdahl P. Elevated serum intercellular adhesion molecule-1 and vascular adhesion molecule-1 among patients with stable angina pectoris who suffer cardiovascular death or non-fatal myocardial infarction. Eur Heart J 1999;20:990-1.
34. Ghaisas NK, Shahi CN, Foley B, Murphy R, Crean P, Walsh M. Elevated levels of circulating soluble adhesion molecules in peripheral blood of patients with Unstable Angina. Am J Cardiol 1997;80:617-9.
35. Mulvihill NT, Foley JB, Murphy RT, Curtin R, Crean PA , Walsh M. Risk stratification in unstable angina and non-Q wave myocardial infarction using adhesion molecules. Heart 2001;85:623-27.
36. Mulvihill NT, Foley JB, Murphy R, Crean P, Walsh M. Evidence of prolonged inflammation in unstable angina and non-Q wave myocardial infarction. J Am Coll Cardiol 2000;36:1210-16.
37. Carlos TM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood 1994;84:2068-2101.
38. Kinlay S, Selwyn AP, Libby P, Ganz P. Inflammation, the endothelium, and the acute coronary syndromes. J Cardiovasc Pharmacol 1998;32:S62-6.
39. Ikeda U, Takahashi M, Shimada K. Monocyte-endothelial cell interaction in atherogenesis and thrombosis. Clin Cardiol 1998;21:11-4.
40. Rothlein R, Mainolfi EA, Czajkowski M, Marlin SD. A form of circulating ICAM-1 in human serum. J Immunol 1991;147:3788-93.
41.Koskinen PK, Lemstrom KB. Adhesion molecule P-selectin and vascular cell adhesion molecule-1 in enhanced heart allograft arteriosclerosis in the rat. Circulation 1997;95:191-6.
42. Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation 1995;92:657-71.
43. Hakkert BC, Kuippers TW, Leeuwenberg JFM, Van Mourik JA, Ross D. Neutrophil and monocyte adherence to and migration across monolayers of cytokine-activated endothelial cells: The contribution of CD18, ELAM-1, VLA-4. Blood 1991;78:2721-26.
44. Bevilacqua MP, Nelson RM. Selectins. J Clin Invest 1993;91:379-87.
45. Zhang RL, Chopp M, Zhang ZG, Phillips ML, Rosenbloom CL, Cruz R, et al. E-selectin in focal cerebral ischemia and reperfusion in the rat. J Cereb Blood Flow Metab 1996;16:1126-36.
46. Lucchesi BR, Werns SW, Fantone JC. The role of the neutrophil and free radicals in ischemic myocardial injury. J Mol Cell Cardiol 1989;21:1241-51.
47. De Servi S, Mazzone A, Ricevuti G, Mazzucchelli I, Fossati G, Gritti D, et al. Clinical and angiographic correlates leukocyte activation in Unstable Angina. JACC 1995;26:1146-50.
48. Siminiak T, Flores NA, Sheridan DJ. Neutrophil interactions with endothelium and platelets possible role in the development of cardiovascular injury. Br Heart J 1995;74:625-30.
49. Hasdai D, Scheinowitz M, Leibovitz E, Sclarovsky S, Eldar M, Barak V. Increased serum concentrations of interleukin -1 in patients with coronary artery disease. Heart 1996;76:24-28.
50. Pober JS, Gimbrone MA. Overlapping patterns of activation of human endothelial cells by interleukin-1, tumor necrosis factor and immune interferon. J Immunol 1986;243:1160-65.
51. Moreno PR, Falk E, Palacios IF, Newell JB, Fuster V, Fallon JT. Macrophage infiltration in acute coronary syndromes: Implications for plaque rupture. Circulation 1994;90:775-78.
52. Jude B, Agraou B, McFadden EP, Susen S, Bauters C, Lepelley P, et al. Evidence for time dependent activation of monocytes in the systemic circulation in unstable angina but not in acute myocardial infarction or in stable angina. Circulation 1994;90:1662-68.
53. Biasucci LM, Vitelli A, Liuzzo G, Attamura S, Caliguri G, Monaco C, et al. Elevated levels of interleukin-6 in unstable angina. Circulation 1996;94:874-77.
54. Buckley CD, Simmons DL. Cell adhesion: A new target for therapy. Mol Med Today 1997;449-56.
55. González-Amaro R, Sánchez- Madrid F. Cell Adhesion molecules: Selectins and Integrins. Crit Rev Immunol 1999;19:389-429.
Recibido: 15 de julio del 2009.
Aprobado: 30 de julio del 2009.
DraC. Consuelo Macías Abraham. Instituto de Hematología e Inmunología. Apartado Postal 8070. Ciudad de La Habana, CP 10800, Cuba. Tel (537) 6438268, 6438695, Fax (537) 6442334. e-mail: ihidir@hemato.sld.cu. Sitio web: http://www.sld.cu/sitios/ihi

