










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter v.26 n.2 Ciudad de la Habana Mayo-ago. 2010
PRESENTACIÓN DE CASOS
Anemia diseritropoyética congénita tipo 1. Presentación de un caso
Type 1 congenital dyserytropoieitc anemia: A case presentation
Dra. Adys I. Gutiérrez DíazI; Dr. Luis G. Ramón RodríguezI; Dra. Daycee Breña EscobarII; Dr. Juan C. Jaime FacundoI; Dr. Jesús Serrano MirabalI; Dr. Alberto Arencibia NúñezI; Dra. Marlen Domínguez ToiracI; Dr. Sergio Machín GarcíaI; Dra. Andrea Menéndez VeitíaI; Dr. Alejandro González OteroI; Prof. Eva SvarchI
IInstituto de Hematología e Inmunología. Ciudad de La Habana, Cuba.
IIHospital Pediátrico Provincial "Eliseo Caamaño". Matanzas, Cuba.
RESUMEN
Las anemias diseritropoyéticas congénitas (ADC) son un grupo de trastornos heridatarios de la hematopoyesis caracterizados por anemia refractaria de severidad variable. Se distinguen 3 tipos fundamentales: 1, 2 y 3. El gen responsable de la ADC-1 (CDAN1) se localiza en el cromosoma 15q15, aunque estudios moleculares recientes evidencian la heterogeneidad de esta enfermedad. Se presenta una paciente de 3 años con diagnóstico de ADC-1 que a los 3 meses de edad comenzó con anemia severa, hiperbilirrubinemia indirecta, reticulocitosis ligera, altos requerimientos transfusionales y alteraciones del desarrollo pondoestatural dado por baja talla. La prueba de Ham fue negativa y en sangre periférica predominó la macrocitosis. En el examen de la médula ósea se observó diseritropoyesis con hiperplasia eritroide, hematopoyesis megaloblástica, precipitados intracitoplasmáticos, núcleos irregulares, cariorrexis, binuclearidad y puentes internucleares. No hubo respuesta al tratamiento con interferón alfa recombinante. La paciente se encuentra con tratamiento quelante con deferroxamina y se ha planteado la posibilidad de un trasplante de células progenitoras hematopoyéticas alogénico no relacionado.
Palabras clave: anemias diseritropoyéticas congénitas, gen CDAN1, interferón alfa.
ABSTRACTS
The congenital dyserytropoietic anemias (CDT) include a series of hematopoiesis hereditary disorders characterized by a refractory anemia of variable severity. There are three fundamental types: 1, 2 and 3. The gen accounted for CDT-1(CDAN1) is located in 15q15 chromosome, although recent studies demonstrate the heterogeneity of this disease. This is the case of a female patient aged 3 diagnosed with CDT-1who at three months old had a severe anemia, indirect hyperbilirubinemia, slight reticulocytosis, high transfusion requirements and stature disorders due to its low height. Ham’s was negative and in peripheral blood there was macrocytosis predominance. Bin bone marrow examination it was possible to observe dyserytropoiesis with erythroid hyperplasia, megaloblast hematopoiesis, intracytoplasm precipitates, irregular nuclei, karyorresis, binuclearization and internuclear bridges. There wasn’t response to treatment with the recombinant type α interferon. Patient is under chelation treatment with deferoxamine and it was proposed the possibility of no-related allogenic of hematopoietic parent cell.
Key words: Congenital dyserytropoietic anemias, CDAN1 gen, type α interferon.
INTRODUCCIÓN
Las anemias diseritropoyéticas congénitas (ADC) son un grupo de desórdenes hereditarios de la hematopoyesis que se caracterizan por anomalías morfológicas de los eritroblastos que conducen a una eritropoyesis ineficaz y anemia secundaria. Las anomalías en las otras líneas celulares no son comunes y se han comunicado en pocos pacientes.1-3 El término fue usado por primera vez por Crookston y otros en casos que después se clasificaron como ADC-24 y por Wendt y Heimpel para casos clasificados posteriormente como ADC-1.5 En 1968 estos 2 últimos autores dividieron las ADC en 3 grupos: tipo 1, 2 y 3, clasificación que aún permanece vigente.1 La ADC-2 también se conoce como HEMPAS, término que identifica una enfermedad hereditaria con eritroblastos multinucleados y positividad para la prueba de suero acidificado conocida como prueba de Ham. En los últimos años se han descrito nuevas formas de ADC basadas en el análisis exhaustivo de los detalles morfológicos, pero su documentación y clasificación son aún imprecisas.1,3,6-8
Según algunos autores, la incidencia de la ADC tipo 1 y 2 es < 1/100 000 nacidos/año.9 A pesar de los esfuerzos realizados para centralizar los casos comunicados, solo se tienen medidas aproximadas de su prevalencia relativa debido a que muchos casos no se diagnostican.1 La ADC-2 es la forma más común, mientras que la ADC-3 es rara.3,7 El resto de las ADC no bien caracterizadas son extremadamente raras7-9 y se han descrito: los tipos IV, IVb, V, VI, VII, VIII y otras variantes diferentes que no pueden clasificarse dentro de estos grupos.1 En este trabajo se presenta a una paciente con ADC-1 diagnosticada en la etapa de lactante.
DESCRIPCIÓN DEL CASO
Paciente femenina de 3 años de edad, mestiza, de procedencia rural, nacida de parto eutócico, a término, normopeso, con antecedentes de una neumonía al mes de nacida y una infección urinaria alta a los 2 meses, para la que requirió tratamiento con amikacina. Durante este último cuadro infeccioso se detectaron cifras de Hb en 81 g/L y por su área de salud se le indicó tratamiento con ácido fólico y fumarato ferroso. A los 3 meses de edad ingresó en el Hospital Pediátrico Provincial "Eliseo Caamaño", Matanzas, Cuba, con astenia y rechazo a los alimentos. Al examen físico se describió palidez marcada de piel y mucosas, polipnea, taquicardia, hepatomegalia de 1 cm y baja talla.
Resultados iniciales de laboratorio:
Hemoglobina 48 g/L; reticulocitos 2,6 %; plaquetas 280 × 109/L; leucocitos 9,6 × 109/L; neutrófilos 35 %; linfocitos 59 %; monocitos 3 %; eosinófilos 3 %.
Bilirrubina total 3,8 mg/dL; bilirrubina indirecta 3,0 mg/dL; bilirrubina directa 0,8 mg/dL; LDH 638,8 UI/L; prueba de Coombs directa negativa; prueba de Ham negativa.
Se interconsultó la paciente con el Instituto de Hematología e Inmunología, donde se realizó estudio a la madre y el padre que dio como resultado electroforesis de hemoglobina AA; resistencia osmótica cualitativa y cuantitativa normales.
En la lámina de sangre periférica se observó anisocitosis, macrocitosis, hipocromía, esferocitos, normoblastos 1 %, punteado básofilo, anillos de Cabot, leucocitos normales y plaquetas adecuadas.
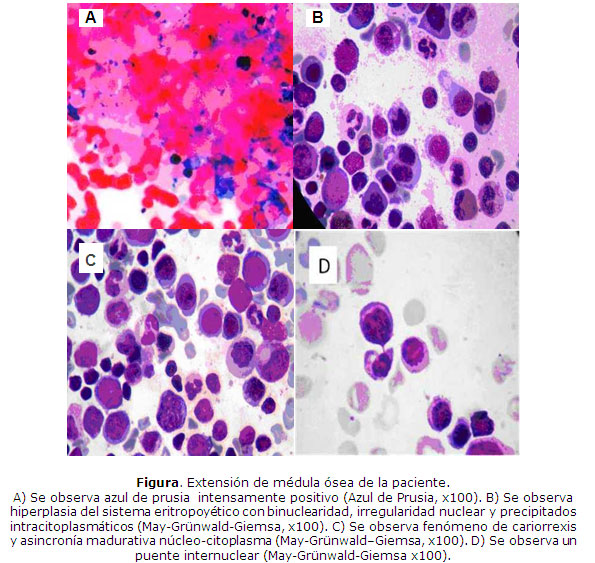
La médula ósea mostró hipercelularidad, azul de prusia intensamente positivo (Fig.A ), sistema megacariopoyético íntegro, sistema granulopoyético con depresión relativa y todas las formas de maduración. El sistema eritropoyético con hiperplasia marcada y todas las formas de maduración, cambios megaloblásticos, asincronía madurativa núcleo-citoplasmática, cariorrexis, núcleos irregulares en forma de hoja de trébol o roseta en las formas maduras, proeritroblastos binucleados, puentes internucleares y precipitados intracitoplasmáticos (Figs. B, C y D).
En la biopsia de médula ósea se observó hipercelularidad, presencia de las 3 series con cambios hiperplásicos eritropoyéticos de ligeros a moderados, inversión de la relación mieloide-eritroide, cambios dismórficos del sistema eritropoyético, apoptosis, retículo grado I/II difuso, y aumento ligero de la hemosiderina.
Se concluyó que la paciente presentaba una ADC-1. Desde su diagnóstico ha presentado requerimientos transfusionales cada 40 a 50 días y hasta el presente ha recibido 31 transfusiones. Se utilizaron sin éxito vitamina B12 e interferón a2b recombinante durante 1 año, en dosis de 3 × 106 U/m2, administrado de forma subcutánea 3 veces a la semana.
En los estudios de HLA realizados a la paciente y a los familiares (padres y hermana) por técnicas serológicas, no se encontraron donantes compatibles para los antígenos de la clase I y II. Recientemente, la paciente presentó cifras de ferritina sérica de 1 691 µg/L, por lo que se inició tratamiento quelante con deferroxamina y se ha planteado la posibilidad de un trasplante no relacionado de progenitores hematopoyéticos.
DISCUSIÓN
La ADC-1 es una entidad rara, pero bien definida, que se ha observado en muchas regiones del mundo. La mayoría de los casos han sido comunicados en países de Europa Central y Occidental, Norte de África y cercano Oriente.2 La forma de herencia es autosómica recesiva y el gen responsable (CDAN1) se localiza en el cromosoma 15 (q15.1-q15.3).2,10 La proteína codificada por este gen se nombra CODANIN1 y su función es desconocida.2,9 El conocimiento más detallado de la ADC-1 requiere una profundización en las funciones de la proteína, su distribución y localización intracelular y la existencia e interacción con otros elementos asociados.11
El conocimiento de la historia natural de la ADC-1 fue posible en parte, por un grupo de casos descritos en varias familias de tribus beduinas de Israel.2 La anemia varía de ligera a severa y los casos más severos se presentan en la infancia, incluso en el período neonatal, y demandan un intenso tratamiento transfusional que en algunos pacientes se comienza intraútero. Los signos de presentación neonatal incluyen hidropis fetal, retardo del crecimiento intrauterino, hepatomegalia, ictericia temprana, incremento de la bilirrubina directa, algunas veces asociada con el síndrome de Gilbert, transaminasas elevadas, persistencia de hipertensión pulmonar en el recién nacido, y la mayoría de los pacientes requieren terapia transfusional, aunque menos del 10 % permanece dependiente de transfusiones.1
En el caso que se presenta, el inicio de los primeros síntomas fue en etapa de lactante con apenas 3 meses de edad y una anemia severa con altos requerimientos transfusionales. En la adolescencia y en edades más avanzadas de la vida se diagnostican aquellos casos con anemia ligera, que es la forma clínica más común. Los hallazgos más frecuentes en el examen físico son: ictericia, esplenomegalia y ocasionalmente hepatomegalia. Se describen también malformaciones congénitas, fundamentalmente anomalías esqueléticas como: dedos supernumerarios, sindactilia, displasia de cadera y baja talla. Además, se ha reportado defecto septal del ventrículo, duplicidad renal, hipoplasia o ausencia de las uñas o de falanges distales en dedos de las manos y pies, metatarsianos adicionales, pie zambo, hoyuelo sacro, hiperterolismo y puente nasal amplio.2,3,7,9,12,13
Los estudios para demostrar alteraciones fenotípicas en nuestra paciente fueron negativos; sin embargo, el retraso en el crecimiento estuvo presente y la anemia severa con ligera hepatomegalia fueron los rasgos distintivos. En 1974, en Cuba se reportó un paciente con una ADC-2 asociada con una enfermedad de von Willebrand. Como rasgo poco común, el hermano presentó una prueba de hemólisis con suero acidificado positiva; no obstante, no se observaron alteraciones morfológicas en la médula ósea.14
En la ADC-1, entre los complementarios se destaca la anemia, que por lo general es macrocítica, con un volumen corpuscular medio (VCM) mayor de 120 fL. Muchas de las alteraciones que se describen en la ADC estuvieron presentes en nuestro caso. En la lámina de sangre periférica se observa, comúnmente, la presencia de macrocitosis, aún en casos en que el VCM está dentro de límites normales; anisocitosis, poiquilocitosis, punteado basófilo y anillos de Cabot, como consecuencia del trastorno madurativo. El número de reticulocitos es normal o ligeramente elevado2,3 y la bilirrubina total está moderadamente aumentada con predominio de la indirecta. La haptoglobina en el suero está disminuida y la LDH incrementada.2,3,12
Los hallazgos más prominentes en esta enfermedad se encuentran en la médula ósea, que se caracteriza por una hipercelularidad a expensas de hiperplasia del sistema eritropoyético. La relación eritroide/mieloide varía entre 3 y 8, comparada con el intervalo normal de 0,2 a 1. Se observan características megaloblastoides en la estructura de la cromatina, signos displásicos que incluyen marcada irregularidad nuclear con núcleos en coliflor, eritroblastos binucleares (particularmente en estadios maduros), algunas veces tri o tetranucleares, principalmente asimétricos (3-7 %), núcleos de diferente tamaño y estructura, y parejas de eritroblastos por lo general maduros, conectados por puentes delgados de cromatina en un intervalo del 0,5 al 8 % de las células. Esta última anomalía es muy difícil de encontrar y se deben examinar al menos 500 eritroblastos. Entre el 30 y 60 % de los eritroblastos se consideran anormales y aparecen con frecuencia los precipitados intracitoplasmáticos (1-15 %) y los fenómenos de cariorrexis (1-5 %).1-3,6,9,12,15 Estas alteraciones se describieron en nuestra paciente y los rasgos distintivos fueron: la irregularidad nuclear y los puentes internucleares. Recientemente se han descrito pacientes portadores de ADC-1 con ausencia de puentes internucleares.16,17
La microscopía electrónica revela una condensación esponjosa de la cromatina con apariencia de queso suizo,18 de la que es responsable la invaginación de la envoltura nuclear, con una localización anómala de los componentes citoplasmáticos dentro del núcleo. Esta característica no está limitada a la ADC-1 y se observa en otros estados diseritropoyéticos.1,3
En la ADC-1, a diferencia de la ADC-2, la prueba de Ham es negativa. El diagnóstico está basado en la presencia de una anemia diseritropoyética congénita inespecífica y la morfología típica de los eritroblastos.2,3,12 La evidencia de eritropoyesis ineficaz en nuestra paciente se basó en la ausencia de reticulocitosis, la hiperplasia eritroide de la médula ósea, el aumento de la bilirrubina indirecta y de la LDH, y la presencia de los cambios morfológicos característicos de esta entidad. Como es característico en la ADC-2, el caso publicado en Cuba por Hernández y otros, presentó multinuclearidad en el 20 % de los eritroblastos y una prueba positiva de hemólisis con suero acidificado.14
Dgany y otros fueron los primeros en demostrar la mutación del gen CDAN, responsable de esta condición.17 Esta mutación se encontró por primera vez en las familias beduinas y se describieron luego en 6 familias no relacionadas con ADC-1 en Europa y otra en Arabia y Polinesia, así como en 8 individuos emparentados en Francia.13 El hallazgo de la mutación en 15 de 16 pacientes en Alemania y Suiza confirmó estos resultados.2 Estas observaciones sugirieron que esta entidad es más que una alteración fenotípica y que la presencia de la mutación del gen CDAN1 fundamenta el diagnóstico de ADC-1. Recientemente se demostró que no todos los pacientes afectados tienen alteraciones del cromosoma 15q15, por lo que debe existir un locus genético alternativo, evidencia de la heterogeneidad genética de esta enfermedad.11
Hasta el momento actual no se ha podido establecer una correlación fenotipo-genotipo en las mutaciones analizadas.2 En 2 hermanos afectados con ADC-1, la severidad clínica fue diferente. Luego de esta evidencia se planteó que es probable que factores que determinan la variabilidad de la eritropoyesis normal influyan más en la expresión clínica que la propia mutación del gen CDAN1.2
El tratamiento de la ADC es esencialmente sintomático e incluye el monitoreo hematológico con transfusiones de sangre según los requerimientos. En estos pacientes, la sobrecarga de hierro es la complicación más frecuente, por lo que es de vital importancia el monitoreo del estado del hierro mediante el estudio de la ferritina sérica y la saturación de la transferrina, así como la estimación del hierro en el hígado mediante biopsia hepática o resonancia magnética nuclear.
Se debe comenzar tratamiento quelante de hierro si la concentración de ferritina sérica se aproxima a niveles de 1 000 µg/L o si hay otra evidencia de daño orgánico secundario a hemocromatosis.2,3,12 El tratamiento con deferroxamina y deferriprona o ambas, según las dosis recomendadas para el tratamiento de hemocromatosis secundaria, es efectivo.2,3,7 Esta complicación no está limitada a los pacientes con requerimientos transfusionales porque la eritropoyesis ineficaz resulta en la disminución de los niveles de hepcidina, por lo que aumenta la absorción del hierro.1
Se ha descrito la asociación de ADC-1 con mutaciones del gen HFE de la hemocromatosis, pero no se ha comprobado un incremento en la severidad de la sobrecarga de hierro. No está claro si mutaciones en otros genes asociados con la hemocromatosis (HJV, HAMP, TFR2, y TMPRSS6), pueden modular este fenómeno. Investigaciones recientes evidenciaron que el GDF15 (un miembro de la súper familia del factor beta transformador del crecimiento que está elevado en la talasemia intermedia), se encuentra también sobreexpresado en la ADC-1. Este factor es capaz de suprimir los niveles de hepcidina, lo que contribuye al aumento de la absorción del hierro.1
Otras complicaciones descritas son las úlceras en los miembros inferiores, la litiasis biliar y la hematopoyesis extramedular, entre otras.1,2
En contraste con la ADC-2, la esplenectomía no se recomienda en la ADC-1 como proceder estándar, aún en los pacientes con anemia marcada, aunque de manera excepcional, en algunos casos severos puede disminuir los requerimientos transfusionales. Al igual que en la ADC-2, la esplenectomía no previene la sobrecarga de hierro.1,2
El primer caso tratado con interferón a fue un paciente con ADC-1 y hepatitis C. Además de corregir las cifras de hemoglobina, se normalizó la absorción enteral de hierro que contribuye a la sobrecarga del mineral en esta enfermedad.19 En otros pacientes se ha utilizado con éxito, lo que sugiere que el interferón a puede resultar importante en su tratamiento.2 En una comunicación reciente, este tratamiento no fue efectivo en 3 pacientes con ADC-1 dependientes de la terapia transfusional. Estas observaciones cuestionan el uso del interferón a en la ADC, por lo que su eficacia debe establecerse con el estudio de un número mayor de pacientes.20
A pesar de estas observaciones, este tratamiento continúa siendo una opción a tener en cuenta en pacientes con requerimientos transfusionales. En nuestra paciente se utilizó durante 1 año sin obtenerse respuesta. Se han administrado formas pegiladas del interferón a en la dosis de 30 µg/semanales, con buenos resultados.1
Aunque la mayoría de los pacientes con ADC-1 son capaces de realizar sus metas sociales y profesionales, otros pueden tener una morbilidad relevante y si no se tratan pueden morir por las secuelas relacionadas con esta enfermedad. Por esta razón, los avances en las investigaciones continúan y en la actualidad el trasplante alogénico de progenitores hematopoyéticos despunta como la única opción terapéutica curativa eficaz en pacientes con ADC-1 dependientes de la terapia transfusional.21 A pesar de todos los intentos por mejorar la condición de nuestra paciente, la transfusión de sangre y el tratamiento quelante de hierrro han devenido la única opción terapéutica. De acuerdo con nuestros conocimientos, este es el segundo caso reportado en Cuba hasta el presente.14
REFERENCIAS BIBLIOGRÁFICAS
1. Renella R, Wood WG. The congenital dyserythropoietic anemias. Hematol Oncol Clin N Am 2009;23:283-306.
2. Heimpel H, Schwarz K, Ebnöther M, Goede JS, Heydrich D, et al. Congenital dyserythropoietic anemia type I (CDA I): Molecular genetics, clinical appearance, and prognosis based on long-term observation. Blood 2006;107:334-40.
3. Marks PW, Glader B. Congenital dyserythropoietic anemias. Wintrobe's Clinical Hematology. 11t ed. Philadelphia: Lippincott Willians and Wilkins Publishers; 2003.
4. Crookston JH, Godwin TF, Wightmann KJR, Dacie JV, Lewis SM, et al. Congenital dyserythropoietic anemia. Abstr XIth Congress Internat Soc Hematol, Sydney; 1966.
5. Wendt F, Heimpel H. Kongenitale dyserythropoetische anaemie bei einem eineiigen Zwillingspaar. Med Klinik 1967;62:172-7.
6. Tamary H, Shalev H , Luria D, Shaft D, Zoldan M, et al. Clinical features and studies of erythropoiesis in Israeli Beduouins with congenital dyserythropoietic anemia type I. Blood 1996;87:1763-70.
7. Lolascon A. Congenital dyserythropoietic anemias: A still unsolved puzzle. Haematologica 2000;85:673-4.
8. Heimpel H. Congenital dyserythropoietic anemias: Epidemiology, clinical significance and progress in understanding their pathogenesis. Ann Hematol 2004;83:613-21.
9. Delaunay J. Congenital dyserythropoietic anemias. Orthanet Encyclopedia, October 2003. http://www.orpha.net/data/patho/GB/uk-CDA.pdf.
10. Tamary H, Shalmon L, Shalev H, Halil A, Shaft D, et al. Localization of the gene for congenital dyserythropoietic anemia type I to chromosome 15q15.1-15.3. Blood 1996;88:144a.
11. Ahmed MR, Chehal A, Zahed L, Taher A, Haidar J, et al. Linkage and mutational analysis of the CDAN1 gene reveals genetic heterogeneity in congenital dyserythropoietic anemia type I (letter). Blood 2006;107:4968-9.
12. Beutler E. The congenital dyserythropoietic anemias. Williams Hematology. 6 ed. Washington: Mc Graw Hill Medical Publisheras; 2001.
13. Tamary H, Dgany O, Proust A, Krasnov T, Avidan N, et al. Clinical and molecular variability in congenital dyserythropoietic anaemia type I. Br J Haemat 2005;130:628-34.
14. Hernández P, Almagro D, Corrala F, Opolski A, Sánchez JA, et al. Association of type II congenital dyserythropoietic anaemia and von Willebrand's disease. Br J Haematol 1974;27:453.
15. Tamary H, Offret H, Dgany O, Foliguet B, Wickramasinghe SN, et al. Congenital dyserythropoietic anaemia, type I, in a Caucasian patient with retinal angioid streaks (homozygous Arg1042Trp mutation in codanin-1). Eur J Haematol 2008;80:271-4.
16. Ru YX, Zhu XF, Zhao SY, Liu JH, Zhong S. Ultrastructural characteristics of congenital dyserythropoietic anemia-type I. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2007;15:117-20.
17. Dgany O, Avidan N, Delaunay J, Krasnov T, Shalmon L, et al. Congenital dyserythropoietic anemia type I is caused by mutations in codanin-1. Am J Hum Genet 2002;71:1467-74.
18. Hodges VM, Molloy GY, Wickramasinghe SN. Genetic heterogeneity of heterogeneity of congenital dyserythropoietic anemia type I. Blood 1999;94:1139-40.
19. Lavabre-Bertrand T, Ramos J, Delfour C, Henry L, Guiraud I, et al. Long-term alpha Interferon treatment is effective on anaemia and significantly reduces iron overload in congenital dyserythropoiesis type I. Eur J Haematol 2004;73:380-3.
20. Marwaha RK, Bansal D, Trehan A, Garewal G. Interferon therapy in congenital dyserythropoietic anemia type I/II. Pediatr Hematol Oncol 2005;22:133-8.
21. Ayas M, Al Jefri A, Baothman A, Al-Mahr M, Mustafa M, et al. Transfusion-dependent congenital dyserythropoietic anemia type I successfully treated with allogeneic stem cell transplantation. Bone Marrow Transplant 2002; 29:681-2.
Recibido: 9 de febrero de 2010.
Aprobado: 24 de febrero de 2010.
Dra. Adys I. Gutiérrez Díaz. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800. Ciudad de La Habana, Cuba. Tel (537) 643 8695, 643 8268, Fax (537) 644 2334. e-mail: ihidir@hemato.sld.cu Website: http://www.sld.cu/sitios/ihi


{kind=link}