










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter v.26 n.3 Ciudad de la Habana sep.-dic. 2010
Hemofilia A adquirida
Acquired hemophilia A
DraC. Delfina Almagro Vázquez
Instituto de Hematología e Inmunología. Ciudad de La Habana, Cuba.
RESUMEN
La hemofilia A adquirida (HAA) es un trastorno hemorrágico poco frecuente caracterizado por la presencia de autoanticuerpos contra el factor VIII (FVIII) circulante. Aproximadamente en la mitad de los casos se ha observado un grupo heterogéneo de procesos patológicos que incluyen, entre otros, enfermedades autoinmunes y malignas y durante el embarazo, parto y puerperio. Las manifestaciones hemorrágicas son variables y fundamentalmente de tipo cutáneo mucoso. El diagnóstico se basa en el hallazgo en un paciente con manifestaciones hemorrágicas, prolongación del tiempo parcial de tromboplastina activado (TPTA), disminución de la actividad del FVIII y presencia de inhibidores del FVIII. El tratamiento de HAA incluye el control de las manifestaciones hemorrágicas y la supresión de la producción del anticuerpo. El concentrado de factor VIIa recombinante (FVIIar) y el concentrado de complejo protrombínico (CCPA) se consideran el tratamiento antihemorrágico de primera línea. Como terapéutica alternativa, en algunos casos puede utilizarse el concentrado de FVIII, la plasmaféresis y la inmunoadsorción extracorpórea. La prednisona sola o asociada con la ciclofosfamida, constituye el tratamiento inmunosupresor de primera línea. En pacientes refractarios puede administrarse como terapéutica de segunda línea, el rituximab (anti-CD20). Con la azatiopina, la ciclosporina, la vincristina y el micofenolato de mofetil, se han obtenido resultados variables.
Palabras clave: hemofilia A adquirida, autoanticuerpos contra el factor VIII, corticosteroides, ciclofosfamida, rituximab.
ABSTRACT
Acquired hemophilia A (AHA) is an uncommon hemorrhagic disorder characterized by presence of autoantibodies to circulating factor VIII. Approximately in half of cases it is noted a heterogeneous group of pathological processes including among others, autoimmune and malignant diseases and during pregnancy, labor and puerperium. Hemorrhagic manifestations are variable and mainly of mucous cutaneous type. Diagnosis is based on the finding of a patient presenting with hemorrhagic manifestations, extension of activated partial thromboplastin time (APTT), decrease of Factor VIII activity, and presence of Factor VIII inhibitors. AHA treatment includes the control of hemorrhagic manifestations and the suppression of antibody production. The recombinant factor VIIIa (rVIIIaF) concentration and the prothrombin-complex concentrations (PCC) are considered like the first-line antihemorrhagic treatment. As alternative therapy in some cases the FCIII concentration, the plasmapheresis and extracorporeal immuno-adsorption may be used. The prednisone alone or associated with cyclophosphamide is the firs-line immunosuppressive treatment. In refractory patients it may be administered as a second-line therapy, the Rituximab (anti-CD20). With the use of Azathioprine, Cyclosporine, Vincristine and the Mycophenolate mofetil variable results have been achieved.
Key words: Acquired hemophilia A, autoantibodies to factor VIII, corticosteroids, Cyclophosphamide, Rituximab.
INTRODUCCIÓN
La hemofilia A adquirida (HAA) es un trastorno hemorrágico poco frecuente caracterizado por la presencia de autoanticuerpos contra el factor VIII (FVIII) circulante. Se ha comunicado una incidencia de alrededor de 1,5 casos por millón por año.1,2 Se presenta con mayor frecuencia en épocas tardías de la vida,3 con un pequeño aumento en mujeres entre 20 y 30 años, asociado con el embarazo, posparto y enfermedades autoinmunes.4 Aproximadamente en la mitad de los casos aparece espontáneamente en individuos sin historia anterior de trastornos hemorrágicos.5
Pavlova y otros han sugerido que estos autoanticuerpos se desarrollan como consecuencia de una interacción compleja de factores genéticos y ambientales, hasta ahora poco conocidos, que provocan un trastorno de la regulación inmune.6 Estos autores demostraron que el polimorfismo del antígeno 4 de los linfocitos T citotóxicos es uno de los múltiples factores genéticos que contribuyen a la aparición de estos autoanticuerpos y que participan también en la modulación de la respuesta autoinmune y en el mantenimiento de la tolerancia periférica.
Los autoanticuerpos contra el FVIII son heterogéneos, particularmente la inmunoglobulina G (IgG), y dependientes de tiempo y temperatura.7 La cinética de la interacción entre el FVIII y este tipo de inhibidor difiere del comportamiento de los aloanticuerpos desarrollados en algunos pacientes con hemofilia hereditaria, ya que muestran una cinética compleja y exponencial que corresponde a un patrón de inactivación tipo 2 con una inhibición incompleta del factor y la presencia de actividad de FVIII residual en el plasma.8
CAUSAS DE LA HEMOFILIA A ADQUIRIDA
Se ha observado que alrededor de la mitad de los pacientes con HAA presentan un grupo heterogéneo de procesos patológicos que incluyen, entre otros, enfermedades autoinmunes y malignas, embarazo, parto y puerperio.9-11 (Tabla 1).

Entre los primeros se encuentran el lupus eritematoso sistémico (LES), la artritis reumatoidea, el síndrome de Sjögren, la miastenia gravis, la esclerosis múltiple y la colitis ulcerativa.12
Dentro de las enfermedades hematológicas malignas los autoanticuerpos contra el FVIII se han presentado en el linfoma no Hodgkin, la leucemia linfocítica crónica, el mieloma múltiple, la macroglobulinemia de Waldenström, la mielofibrosis y los síndromes mielodisplásticos.13
Los tumores sólidos en los que con mayor frecuencia se ha observado la HAA son: carcinoma de próstata, mama, pulmón, estómago, páncreas, colon, cabeza, cuello y riñón. Aunque la desaparición del inhibidor parece estar relacionada con la evolución satisfactoria del tumor, su reaparición no se considera un marcador adecuado de la recurrencia del proceso maligno.14
Los inhibidores del FVIII se han encontrado también en algunos procesos alérgicos como el asma, y en reacciones medicamentosas, en particular a las sulfonamidas; y con menor frecuencia, a la penicilina, fenitoína, cloranfenicol, metildopa y alfa-interferón, así como en enfermedades dermatológicas tales como el pénfigo, la psoriasis y después de la administración de vacunas.12
La llamada HAA post partum es una complicación poco frecuente, pero habitualmente grave, asociada con el embarazo, parto y puerperio. En la mayoría de los casos se presenta en mujeres primigrávidas.
Aunque las manifestaciones hemorrágicas pueden aparecer hasta 2 meses después del parto, con mayor frecuencia ocurren sangramientos severos durante el trabajo de parto, parto y puerperio inmediato, que en ocasiones requieren histerectomía, por lo que deben ser tratadas de manera inmediata.15,16
El inhibidor de FVIII post partum tiene la tendencia a desaparecer espontáneamente en un número importante de casos en el curso de meses, es de buen pronóstico y la mortalidad es baja comparado con otras causas de HAA.12,17,18
La recurrencia del inhibidor en ulteriores embarazos es raramente observada;19,20 su ocurrencia podría provocar sangramiento fetal importante por la transferencia trasplacentaria del autoanticuerpo.
Es necesario señalar que ante la presencia de una hemorragia posparto en que no se halle una causa obstétrica, debe investigarse la presencia de una HAA.
Se ha observado que en los inhibidores del FVIII post partum los corticosteroides tienen mayor eficacia en la erradicación del autoanticuerpo que en el resto de los procesos etiológicos con esta complicación.
El manejo de los episodios hemorrágicos es similar, como se verá más adelante.21,22
MANIFESTACIONES CLÍNICAS
Las manifestaciones hemorrágicas en la HAA son variables y se presentan en individuos sin historia previa de coagulopatía:3
Sangramientos cutáneo-mucosos.
- Equímosis.
- Epístaxis.
- Menorragia.
Hematuria.
Sangramiento gastrointestinal.
Sangramientos infrecuentes.
- Hemartrosis.
- Hematomas musculares.
De manera diferente a lo que ocurre en la hemofilia hereditaria, el nivel plasmático del FVIII no es un predictor adecuado del riesgo de episodios hemorrágicos23 y estos son fundamentalmente del tipo cutáneo-mucoso (equímosis, epístaxis, menorragia, hematuria, sangramiento digestivo); los hematomas musculares y las hemartrosis son poco comunes.4 Ocasionalmente pueden ocurrir hemorragias sistémicas con peligro para la vida del paciente.
Collins y otros encontraron una incidencia del 9 % de sangramientos fatales en pacientes con HAA.1 Estos mismos autores hallaron que la causa de muerte en las primeras semanas se debía principalmente a sangramientos digestivos y pulmonares, y las muertes tardías fueron consecutivas a hemorragia intracraneal y retroperitoneal, así como a complicaciones infecciosas secundarias al tratamiento inmunosupresor.
DIAGNÓSTICO
El diagnóstico de la HAA debe sospecharse en un paciente con manifestaciones hemorrágicas de comienzo agudo sin historia previa de trastornos de la hemostasia, que se acompaña de una prolongación inexplicable del tiempo parcial de tromboplastina activado (TPTA).24
Determinan el diagnóstico de la hemofilia A adquirida:
- La prolongación del tiempo parcial de tromboplastina activado en mezclas de plasma del paciente y plasma normal.
- La disminución de la actividad del factor VIII.
- La presencia de inhibidores del factor VIII.
La prolongación del TPTA en una mezcla de plasma del paciente y plasma normal después de 2 horas de incubación a 37 °C, denota la presencia de un inhibidor.25 Sin embargo, es necesaria la determinación de los factores VIII, IX y XI para un diagnóstico adecuado. El hallazgo de una disminución aislada de la actividad de FVIII sugiere el diagnóstico de HAA. Es necesario tener en cuenta que en algunos casos, todos los factores del sistema intrínseco pueden estar disminuidos como consecuencia de la neutralización por el inhibidor del factor VIII presente en los plasmas sustratos utilizados para las determinaciones de estos factores en el plasma.26
La presencia del anticoagulante lúpico (AL) es otro elemento que puede interferir en la determinación del TPTA, de la actividad del FVIII y de los inhibidores, por su conocida acción sobre los fosfolípidos utilizados en estos ensayos. Aunque desde el punto de vista clínico el AL no está asociado con manifestaciones hemorrágicas y se caracteriza por una tendencia a la hipercoagulabilidad27 y, además, puede diferenciarse por los resultados del estudio de mezcla del plasma del paciente y plasma normal, ya que a diferencia de los inhibidores del FVIII la neutralización del factor no se incrementa con la incubación de la mezcla,28 en caso de sospecha de AL debe realizarse su determinación específica.29
La determinación del inhibidor del FVIII es imprescindible para establecer el diagnóstico de HAA. El método desarrollado por Kasper y otros,7 utilizado habitualmente, fue diseñado para cuantificar aloanticuerpos contra el FVIII que muestran una cinética tipo 1, como ocurre en la hemofilia A hereditaria. En el caso de los autoanticuerpos en la HAA, por sus características cinéticas ya mencionadas, provocan una inhibición incompleta del FVIII con actividad del factor en el plasma, aún en presencia de altos títulos del inhibidor, por lo que esta técnica, aunque detecta el autoanticuerpo, no nos permite establecer su verdadera potencia.28
TRATAMIENTO
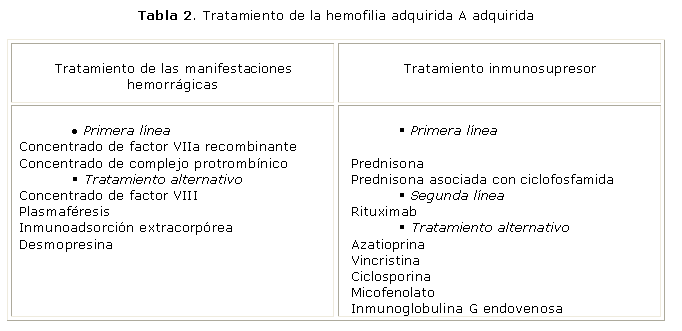
El tratamiento de la HAA incluye 2 aspectos fundamentales: el control de las manifestaciones hemorrágicas y la supresión de la producción del autoanticuerpo (tabla 2).
El tratamiento antihemorrágico debe iniciarse de manera inmediata independientemente del título del inhibidor.1,30 En esta etapa es necesario evitar los procederes invasivos siempre que sea posible.
Los productos que sortean el efecto del inhibidor: el concentrado de factor VIIa recombinante (FVIIar) y el concentrado de complejo protrombínico (CCPA), se consideran la terapéutica de primera línea para detener el sangramiento en pacientes con HAA.23,31
El uso exitoso del FVIIar en pacientes con inhibidores del FVIII comenzó en la década de los 80 en pacientes con hemofilia A hereditaria.32 En los últimos años se ha demostrado su eficacia como agente hemostático en otros trastornos hemorrágicos33,34 que incluyen la HAA.35,36 Se administra a la dosis de 90-120 µg/kg cada 2-3 horas hasta obtener la hemostasia que ocurre habitualmente entre 24 y 72 horas, en dependencia del sitio y la severidad del sangramiento.37
El FVIIar es un agente con potente actividad procoagulante, por lo que teóricamente su uso podría representar un riesgo potencial para el desarrollo de eventos trombóticos. Sumner y otros encontraron en 189 pacientes tratados con FVIIIar, 7,5 % de accidentes trombóticos, particularmente arteriales.35 Sin embargo, se desconoce si algunos factores de riesgo como edad avanzada y enfermedades cardiovasculares, entre otras, tienen alguna influencia en estos resultados. Por otra parte, en una revisión reciente Abshire y otros hallaron menos del 1 % de eventos trombóticos en 800 000 infusiones de FVIIar en pacientes con hemofilia A hereditaria y adquirida.37 En general, se considera que la incidencia de complicaciones trombóticas en pacientes tratados con FVIIar es baja y que el uso de este producto es seguro.
El CCPA, que es un concentrado plasmático de factores dependientes de la vitamina K, ha sido ampliamente utilizado en pacientes con hemofilia A hereditaria e inhibidores del FVIII y en menor medida en la HAA. Forma parte, junto con el FVIIar, del tratamiento de primera línea en estos pacientes. Con dosis de 50-100 UI/kg cada 8-12 horas se han obtenido respuestas favorables.31 En algunos casos se han hallado complicaciones trombóticas, particularmente cuando se administra en dosis mayores de 200 UI/kg, incluida la coagulación intravascular diseminada (CID).30
La asociación de agentes fibrinolíticos con estos productos ha sido muy debatida, aunque algunos autores no recomiendan esta asociación: Baudo y otros utilizan el tratamiento con FVIIar y ácido tranexámico de modo rutinario, particularmente en presencia de sangramientos mucosos.38
En el caso de no estar disponible el tratamiento de primera línea con FVIIar o CCPA, pueden utilizarse otras medidas terapéuticas alternativas para el control de los episodios hemorrágicos, que en determinadas circunstancias pueden ser efectivos.
De manera similar a lo que ocurre con la hemofilia A hereditaria, el concentrado de FVIII plasmático o recombinante solo sería eficaz en aquellos pacientes con una tasa de inhibidor menor de 5 UB/mL, ya que un nivel mayor del anticuerpo inactiva el factor transfundido.30
La plasmaféresis puede ser utilizada en situaciones de urgencia cuando sea necesaria la eliminación rápida del inhibidor;39 habitualmente de 5 a 7 sesiones en equipo de aféresis son suficientes.
Con este mismo objetivo puede aplicarse la inmunoadsorción extracorpórea, que se considera una técnica útil y segura para la eliminación del inhibidor.40,41 No obstante, este proceder requiere personal especializado y un buen acceso venoso.
El uso de la desmopresina (DDAVP) ha sido sugerido en algunos casos; sin embargo, su efectividad no está claramente demostrada. Tiene las mismas limitaciones del concentrado de FVIII, de manera que solo estaría indicada en pacientes con sangramientos leves y bajo título del autoanticuerpo.42,43 Para valorar su indicación en pacientes ancianos es necesario tener en cuenta los efectos secundarios de este medicamento.
El tratamiento inmunosupresor es el otro elemento esencial del manejo terapéutico de pacientes con HAA y de manera similar al control de los episodios hemorrágicos, debe iniciarse inmediatamente después de realizado el diagnóstico.
El nivel del inhibidor no es un predictor del riesgo hemorrágico; sin embargo, está relacionado con la respuesta al tratamiento inmunosupresor y se ha observado que aquellos pacientes con un título más bajo del autoanticuerpo responden mejor al tratamiento.30
El régimen de inmunosupresión que ha mostrado mayor eficacia en la supresión de la producción del inhibidor es la administración de prednisona en dosis de 1mg/kg/día durante 4-6 semanas, sola o asociada con ciclofosfamida 1,5 a 2 mg/kg/día durante 6 semanas.15,23,44,45 Algunos autores han encontrado mejores resultados con esta asociación. 1 No obstante, el estudio de supervivencia global y libre de enfermedad no muestra diferencia entre el tratamiento con prednisona sola y su combinación con la terapia citotóxica.30
Recientemente se ha introducido un nuevo agente terapéutico en el tratamiento de la HAA, el rituximab,46-49 un anticuerpo monoclonal quimérico anti-CD20 que se utiliza en el tratamiento del linfoma.50 Por su capacidad para depletar los linfocitos B productores de autoanticuerpos51 se ha administrado en varias enfermedades autoinmunes.52-55 La dosis habitual es de 375 mg/m2/ semana hasta 4 semanas. Se considera de utilidad como terapéutica de segunda línea en pacientes refractarios al tratamiento con corticosteroides o a su asociación con ciclofosfamida; o cuando esté contraindicado este régimen terapéutico.
Con el uso de otros inmunosupresores como terapia alternativa se han obtenido respuestas variables. Entre las más utilizadas se encuentran la azatioprina, la vincristina, la ciclosporina y el micofenolato de mofetil.10,30,56,57
La mayoría de las drogas inmunosupresoras están asociadas con efectos secundarios, en ocasiones de gravedad, como en el caso de sepsis que pueden poner el peligro la vida del paciente, 15,58 de manera que sería conveniente realizar un análisis previo de las características del enfermo y su posible beneficio y seguridad antes de iniciar el tratamiento con determinado inmunosupresor.
Los resultados del tratamiento con IgG endovenosa en altas dosis en pacientes con HAA, han sido en general pobres.10,15 Schwartz y otros solo encontraron una supresión del inhibidor en el 10 % de los casos con bajo título del anticuerpo.59 Algunos autores no recomiendan su uso en estos pacientes.30
Aunque teóricamente podría estar indicada en algunos enfermos, no se ha definido aún si la aplicación de regímenes de inmunotolerancia similares a los utilizados en pacientes con hemofilia A hereditaria e inhibidores, son de utilidad en estos casos.
Se considera que el paciente ha alcanzado la remisión completa cuando desaparece el inhibidor y el FVIII se encuentra dentro de límites normales.
Huth-Kühne y otros han recomendado el seguimiento ulterior de estos enfermos con TPTA y determinación de la actividad del FVIII, mensualmente durante los primeros 6 meses; cada 2 ó 3 meses hasta 12 meses; y cada 6 meses durante el segundo año.30
REFERENCIAS BIBLIOGRÁFICAS
1. Collins PW, Hirsch S, Baglin TP, Dolan G, Hanley J, Makris M, et al. Acquired hemophilia A in the United Kingdom. A 2 year National Surveillance study by the United Kingdom Haemophilia Centre Doctors'organization. Blood 2007;109:1870-7.
2. Collins PW, Mccartney N, Davies R, Lees S, Giddings J, Majer R. A population based unselected, consecutive cohort of patients with acquired haemophilia A. Br J Haematol 2004;124:86-90.
3. Green D. The management of acquired haemophilia. Haemophilia 2006;12(Suppl 5):32-6.
4. Yee TT, Taher A, Pasi KJ, Lee CA. A survey of patients with acquired hemophilia in a hemophilia centre over 28-year-period. Clin Lab Haematol 2000;22:275-8.
5. Cohen AJ, Kessler CM. Acquired inihibitors. Baillieres Clin Haematol 1996;9:331-54.
6. Pavlova A, Díaz-Lacava A, Zeitter H, Satoquina J, Niemann B, Krausen M, et al. Increased frequency of the CTLA-4 49 A/E polymorphism in patients with acquired haemophilia A compared to healthy controls. Haemophilia 2008;14:355-60.
7. Kasper CK, Aledort LM, Aronson L, Counts RB, Edson JR, Van Eys J, et al. A more uniform measurement of factor VIII inihibitors. Thromb Diath Haemorr 1975;34:869-72.
8. Lechner K. Acquired inhibitors in non-hemophilic patients. Haemostasis 1974;3:65-93.
9. Franchini M, Gandini G, DiPaulantonio T, Mariani G. Acquired hemophilia A: A concise review. Am J Haematol 2005;80:55-63.
10. Di Bona E, Schieroni M, Castaman G, Ciarerella N, Rodeghiero F. Acquired haemophilia: Experience of two Italian Centres with 17 new cases. Haemophilia 1997;3:183-8.
11. Hutin MB. Acquired inhibitors in malignant and non malignant disease state. Am J Med 1991;91:9-13.
12. Kessler CM, Acs P, Mariani G. Acquired disorders of coagulation. The immune coagulopathies. En: Colman RW, Marder VJ, Clowes AW, George JN, Goldhaber SZ, eds. Hemostasis and thrombosis. Basic Principles and Clinical Practice. 5 ed. Philadelphia: Lippincott, Williams and Wilkins; 2006. pp. 1061-84.
13. Sallah S, Nguien NP, Abdallah JM. Acquired hemophilia in patients with hematologic malignancies. Arch Pathol Lab Med 2004;124:730-4.
14. Sallah S, Wan JY. Inhibitors against factor VIII in patients with cancer. Analysis of 41 patients. Cancer 2001;91:1067-74.
15. Delgado J, Yiménez-Yuste V, Hernández Navarro, Villar A. Acquired haemophilia: Review and meta-analysis focused on therapy and prognostic factors. Br J Haematol 2003;121:21-35.
16. Michiels J. Acquired hemophilia in women post partum: Clinical manifestations, diagnosis and treatment. Clin Appl Thromb Hemost 2000;6:82-6.
17. Santoro RC, Prejano S. Postpartum acquired hemophilia A: A description of three cases and literature review. Blood Coagulat Fibrinolysis 2009;20:461-5.
18. Hauser I, Schneider B, Lechner K. Post partum factor VIII inihibitors. A review of the literature with special reference to the value of steroid and immunosuppresive treatment. Thromb Haemost 1995;73:1-15.
19. Baudo F, de Cataldo F. Acquired factor VIII inhibitors in pregnancy; data from the Italian Haemophilia Register relevant to clinical practice. Br J Obstet Gynecol 2003;110:311-4.
20. Solymoss S. Postpartum acquired factor VIII inhibitors: Results of a survey. Am J Hematol 1998;59:1-4.
21. Bona S, Hantoushzadeh S. Acquired hemophilia as a cause of primary post-partum hemorrhage. Acta Iran Med 2007;10:107-10.
22. Rashyap R, Choudhry VP, Mahapatra M, Chumber S, Saxena R, Kaul HL. Postpartum acquired haemophilia clinical recognition and management. Haemophilia 2001;7:327-30.
23. Collins PW, Budde V, Rand JH, Federici AB, Kessler CM. Epidemiology and general guidelines of the management of acquired haemophilia and von Willebrand syndrome. Haemophilia 2008;14:49-55.
24. Mashiko R, Yamamoto T, Sato M, Noguchi S, Matsumura A. Acquired hemophilia first manifesting as life-threatening intracranial hemorrhage Case Report Neurol Med Chir 2009;49:93-5.
25. Lossing TS, Kasper CK, Feinstein DJ. Detection of factor VIII inhibitors with the partial thromboplastin time. Blood 1977;49:793-7.
26. Kasper CK. Laboratory tests for factor VIII inhibitors their variation, significance and interpretation. Blood Coagul Fibrinolysis 1991;2(Suppl 1):7-10.
27. Simioni P, Prandoni P, Zanon E, Saracino MA, Scudeller A, Villalta S, et al. Deep venous thrombosis and lupus anticoagulant. A case-control study. Thromb Haemost 1996;76:187-9.
28. Cawryl MS, Hoyer IW. Inactivation of factor VIII coagulant activity by two different types of human antibodies. Blood 1982;60:1103-9.
29. Greaves M, Cohen H, Machon SJ, Mackie I. Guidelines on the investigation and management of the anti-phospholipid syndrome. Br J Haematol 2000;109:704-15.
30. Huth- Kühne A, Baudo F, Collins P, Ingerslev J, Kessler CM, Lévesque H, et al. International recommendations on the diagnosis and treatment of patients with acquired hemophilia A. Haematologica 2009;94:566-75.
31. Sallah S. Treatment of acquired haemophilia with factor eight inhibitor bypassing activity. Haemophilia 2004; 10:169-73.
32. Hedner V, Glazer S, Pingel K, Alberts KA, Blomback M, Schulman S, et al. Successful use of recombinant factor VIIa in a patient with severe hemophilia A during synovectomy. Lancet 1988;2:1193.
33. Almagro D. Uso del factor VII activado recombinante como agente hemostático en trastornos hemorrágicos. Rev Cub Hematol Inmunol Hemoter 2010;26(2).
34. Roberts HR, Monroe DM, White GC. The use of recombinant factor VIIa in the treatment of bleeding disorders. Blood 2004;104:3858-64.
35. Sumner MJ, Geldziler BD, Pedersen M, Seremetis S. Treatment of acquired haemophilia with recombinant activated FVII: A critical appraisal. Haemophilia 2007;13:451-61.
36. Hay CR, Negrier C, Ludlam CA. The treatment of bleeding in acquired haemophilia with recombinant factor VIIa: A multicentre study. Thromb Haemost 1997;78:1463-7.
37. Abshire T, Kenet G. Recombinant factor VIIa: Review of efficacy, dosing regimens and safety in patients with congenital and acquired factor VIII or IX inhibitors. J Thromb Haemost 2004;2:899-909.
38. Baudo F, de Cataldo F. Acquired hemophilia; A critical bleeding syndrome. Haematologica 2004;89:96-100.
39. Erskine JG, Bumet AK, Walker ID, Davidson JE. Plasma exchange in non-haemophiliac patients with inhibitors to factor VIIIC. Br Med J 1981;283:760.
40. Huth-Kühne A, Lages P, Hampel H, Zimmermann R. Management of severe hemorrhage and inhibitor elimination in acquired hemophilia. The modified Heidelberg-Malmo protocol. Hamatologica 2003;88:86-92.
41. Franchini M, Sassi M, Dell'Anna P, Manzato F, Saluagno GL, Montagnana M, et al. Extracorporeal immunoadsorption for the treatment of coagulation inhibitors. Semin Thromb Hemost 2009;35:76-80.
42. Muhm M, Grois N, Kier P, Stumpflen A, Kyrle P, Pabinger I, et al. 1-Deamino-8-D-arginine vasopressin in the treatment of non-haemophilic patients with acquired factor VIII inhibitor. Haemostasis 1990;20:15-20.
43. Mudad R, Kane WH. DDAVP in acquired hemophilia A: Case report and review of the literature. Am J Hematol 1993;43:295-9.
44. Green D. Cytotoxic suppression of acquired factor VIII: C inhibitors. Am J Med 1991;91:14S-19S.
45. Green D, Rademarker AW, Briet E. A prospective, randomized trial of prednisone and cyclophosphamide in the treatment of patients with factor VIII autoantibodies. Thromb Haemost 1993;70:753-7.
46. Ichikawa S, Kohata K, Okitsu Y, Suzuki M, Nakajima S, Yamada MF, et al. Acquired hemophilia A with sigmoid colon cancer: Successful treatment with Rituximab followed by sigmoidectomy. Int J Hematol 2009;90:33-6.
47. Mei-Dan, Walfisch A, Martinowitz V, Hallak M. A rapidly progressive life-threatening postpartum hemorrhage: Successful treatment with anti-CD20 monoclonal antibody. Obstet Gynecol 2009;114:417-9.
48. Aggarwal A, Grewal R, Green RJ, Boggio L, Green D, Weksler BB, et al. Rituximab for autoimmune haemophilia: A proposed treatment algorithm. Haemophilia 2005;11:13-9.
49. Franchini M. Rituximab in the treatment of adult acquired hemophilia A: A systematic review. Crit Rev Oncol Hematol 2007;63:47-52.
50. Coiffier B, Lepage E, Briere J, Herbrecht R, Tilly H, Bouabdallah R, et al. Chop chemotherapy plus Rituximab compared with chop alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 2002;346:235-42.
51. Kimby E. Tolerability and safety of Rituximab (Mabthera). Cancer Treat Rev 2005;31:456-73.
52. Sibilia J, Sordet C. Rituximab: An original biotherapy in auto-immune disorders. Rev Med Interne 2005;26:485-500.
53. Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, et al. Efficacy of B-cell-targeted therapy with Rituximab in patients with rheumatoid arthritis. N Engl J Med 2004;350:2572-81.
54. Leandro MJ, Cambridge G, Edwards JC, Ehrenstein MR, Isenberg DA. B-cell depletion in the treatment of patients with systemic lupus erythematosus: A longitudinal analysis of 24 patients. Rheumatology 2005;44:1542-5.
55. Arnold DM, Pentali F, Crowther MA, Meyer RM, Cook RJ, Sigovin C, et al. Systematic review, efficacy and safety of Rituximab for adults with idiopathic thrombocytopenic purpura. Ann Inter Med 2007;146:25-33.
56. Lian ECY, Larcada A, Chiu AYZ. Combination immunosuppressive therapy after factor VIII infusion for acquired factor VIII inhibitor. Ann Intern Med 1989;110:774-8.
57. Eisert S, Mosler K, Laws HJ, Gobel V. Successful use of mycophenolate mofetil and prednisone in a 14 year-old girl with acquired hemophilia A. Thromb Haemost 2005;93:792-3.
58. Yemanovchi J, Abe T, Azuma T, Narumo H, Fujiwara H, Yakushijin Y, et al. Acquired hemophilia complicated with multiple muscle abscess by Nocardia. Rirsho Ketsueki 2009;50:495-8.
59. Schwartz RS, Gabriel DA, Aledort LM, Green D, Kessler CM. A prospective study of treatment of acquired (autoimmune) factor VIII inhibitors with high-dose intravenous gammaglobulin. Blood 1995;86:797-804.
Recibido: 15 de marzo del 2010.
Aprobado: 2 de abril del 2010.
Prof. Delfina Almagro Vázquez. Instituto de Hematología e Inmunología. Apartado 8070, Ciudad de La Habana, CP 10800, Cuba. Tel (537) 643 8695, 8268, Fax (537) 644 2334. e-mail: ihidir@hemato.sld.cu
Website: http://www.sld.cu/sitios/ihi


{kind=link}