










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter v.26 n.3 Ciudad de la Habana sep.-dic. 2010
PRESENTACIÓN DE CASOS
Linfohistiocitosis hemofagocítica. Informe de un caso
Hemophagocytic lymphohistiocytosis: A case report
Dr. Juan Carlos Jaime FagundoI; Dra. Lisette Trelles PorroII; Dra. Gricel Durán GuarchII; Lic. Juana Luisa Rodríguez PérezII; Dra. Adys Gutiérrez DíazI; Dra. Valia Pavón MoránI; Dr. Alejandro González OteroI
IInstituto de Hematología e Inmunología. Ciudad de La Habana, Cuba.
IIHospital Pediátrico Docente "William Soler". Ciudad de La Habana, Cuba.
RESUMEN
El síndrome hemofagocítico es un desorden del sistema fagocítico-mononuclear que se caracteriza por una proliferación histiocítica generalizada y benigna con fenómeno de hemofagocitosis. Se presenta un caso de linfohistiocitosis hemofagocítica en un recién nacido de 17 días, con hepatoesplenomegalia, pancitopenia y serología positiva para citomegalovirus. Se hace el diagnóstico; se trata con protocolo para linfohistiocitosis hemofagocítica, con remisión completa del paciente.
Palabras clave: hemofagocitosis, citomegalovirus, linfohistiocitosis.
ABSTRACT
The hemophagocytosis syndrome is a disorder of the phagocytic-mononuclear system characterized by a systemic and benign histiocytic proliferation with hemophagocytosis phenomenon. This is a case diagnosed with a hemophagocytic lymphohistiocytosis in a newborn aged 17 presenting with hepatosplenomegaly, pancytopenia and cytomegalovirus-positive serology. Diagnosis is made; is treated with hemophagocytic lymphohistiocytosis protocol a total remission of patient.
Key words: Hemophagocytosis, cytomegalovirus, lymphohistiocytosis.
INTRODUCCIÓN
El término "histiocitosis hemofagocítica" se considera una entidad rara y tan poco frecuente que muchas veces pasa inadvertida para los pediatras y neonatólogos y en ocasiones, los cuadros son diagnosticados como una coagulación intravascular diseminada (CID).
Los síndromes hemofagocíticos o histiocitosis hemofagocíticas, son cuadros poco conocidos, con presencia de células de tipo macrofágico en diferentes órganos y tejidos como hígado, bazo, medula ósea u otros, donde el hallazgo característico es la presencia de hemofagocitosis. Su importancia radica en su gravedad, pues son cuadros muy agresivos que evolucionan en poco tiempo de forma fulminante, con un fallo multiorgánico letal, antes de establecer un diagnóstico.1
Dentro de los síndromes hemofagocíticos se distinguen 2 cuadros que se diferencian por su forma de presentación. La linfohistiocitosis hemofagocítica familiar, que es una enfermedad rara, descrita en 1952 por Farquhar y Claireaux,2 a la que llamaron reticulosis familiar hemofagocítica. La incidencia anual se estima en uno por millón de niños; en el 80 % de los casos, la edad de comienzo es generalmente inferior a los 2 años y el 65 % ocurre en menores de 6 meses.2 Es algo más frecuente en varones. La consanguinidad, en muchos casos, hace suponer la posibilidad de una herencia autosómica recesiva, aunque el 25 % de los pacientes no son familiares y existen defectos en el gen de perforina.3
En las formas secundarias, la primera reseña corresponde a Risdall y otros,4 quienes en 1979 describieron un síndrome clínico caracterizado por proliferación de histiocitos no neoplásicos con fenómeno de hemofagocitosis, asociado con infección por virus, y que llamaron síndrome hemofagocítico asociado a virus. Posteriormente, este síndrome se ha descrito asociado con todo tipo de infecciones y con enfermedad no infecciosa que produzca estrés inmunológico, como linfomas, leucemias, síndromes mielodisplásicos, carcinomas, entre otros. También se ha relacionado con transfusiones recientes de hemocomponentes y con alimentación parenteral prolongada que incluya lípidos solubles. En conjunto, se habla de síndromes hemofagocíticos reactivos o secundarios.5
En el año 2004 se revisaron los criterios diagnósticos de la linfohistiocitosis hemofagocítica:6, 7
A. Enfermedad familiar/ defecto genético conocido.
B. Criterios clínicos y de laboratorio (5 de 8 criterios).
• Fiebre.
• Esplenomegalia.
• Citopenias: 2 o más líneas celulares.
• Hemoglobina menor de 90 g/L.
• Plaquetas menor de 100 X 109/L.
• Neutrófilos menor de 1 X109/L.
• Hipertrigliceridemia más de 3mmol/L, hipofibrinogenemia menor de 1,5 g/L o ambos.
• Ferritina ³;500 µg/L.
• CD 25 soluble ³;2400 uds/m3.
• Disminución o ausencia de la actividad de las células NK.
• Hemofagocitosis en médula ósea, LCR o ganglios.
• Para el diagnóstico se necesita la comprobación del defecto genético ó 5 de los 8 criterios propuestos.
PRESENTACIÓN DEL CASO
Paciente masculino atendido en el Servicio de Neonatología del Hospital Pediátrico Docente "William Soler" (HPWS), de 17 días de nacido, con fiebre, manifestaciones hemorrágicas, hepatoesplenomegalia, palidez cutáneo mucosa marcada y confirmación de infección bacteriana por estafilococo, para lo que llevó tratamiento con ceftriaxona y amikacina.
Complementarios realizados
Hemograma: Hb 66 g/L; leucocitos 1,3 x 109/L; polimorfonucleares 0,09; linfocitos 0,81; monocitos 0,04; eosinófilos 0,06; conteo de plaquetas 10 X 109/L.
Glucosa: 4,0 mmol/L.
Creatinina: 73 mmol/ L.
Aspartatoaminotransferasa (TGO) 68 U/L; alaninoaminotransferasa (TGP) 54 U/L; lactato deshidrogenasa (LDH) 728 U/L.
Tiempo de protrombina: control 13", paciente 36"; tiempo parcial de tromboplastina (TPT Kaolín): control 33", paciente 68".
Fibrinógeno: 1,27 g/ L.
Triglicéridos: 1,92 mmol/L.
Serología para VIH: negativa.
Serología para EBV: negativa.
Serología para CMV: (IgM) positiva.
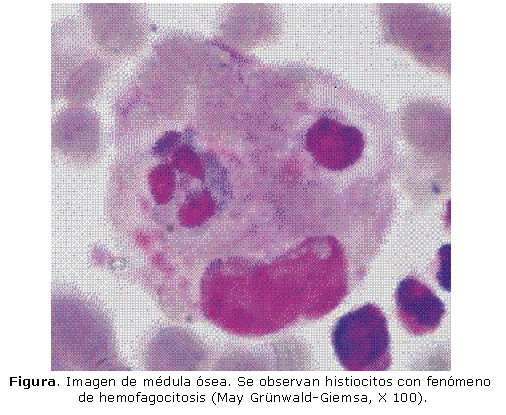
El Servicio de Neonatología solicitó interconsulta con nuestro Instituto de Hematología e Inmunología (IHI), donde se le realizó hemograma, por el que se comprobó la pancitopenia. Se realizó medulograma, en el que se observó la infiltración medular por histiocitos con fenómeno de hemofagocitosis (fig.) y se observó la afectación de las 3 líneas hematopoyéticas. Además, se comprobó hipofibrinogenemia.
Se inició tratamiento con inmunoglobulina G (Intacglobin ®, IMEFA, Cuba) 400mg/kg/día durante 5 días; y dexametasona (DXT) 10 mg /m2/día por vía endovenosa. Tres días después se inició el protocolo para linfohistiocitosis hemofagocítica 2004 (HLH 2004), que incluye DXT, ciclosporina y etopósido.6 Se apoyó con plasma fresco congelado y plasma rico en plaquetas, así como con transfusión de glóbulos, según necesidades.
Se valoró por infectología. En los estudios realizados se encontró serología positiva para infección activa por citomegalovirus , lo que pudo ser el desencadenante del cuadro, por lo que se inició terapia con ganciclovir en las dosis habituales, la que se suspendió al segundo día por leucopenia y trombocitopenia severas.
El paciente tuvo cuadro de sangramiento digestivo bajo por posible afección intestinal, lo que llevó a la aplicación de sonda de Levine y reajustes en su alimentación, la que se inició de forma parenteral.
A los siete días de iniciada la terapia comenzó la mejoría del cuadro clínico con la disminución paulatina de las manifestaciones hemorrágicas y la desaparición de la hepatoesplenomegalia, con normalización del hemograma y el coagulograma y del cuadro neurológico. Se mantuvo bajo el tratamiento con las 3 drogas hasta la octava semana, según protocolo para los casos que han tenido remisión en ese momento del tratamiento, en que se comenzó a disminuir la dosis DXT hasta suspender. El paciente se ha seguido por consulta durante seis meses, en los que se ha mantenido asintomático con hemogramas normales y sin evidencia de reactivación de la enfermedad.
DISCUSIÓN
Consideramos que en este caso se actuó rápidamente por el Servicio de Neonatología del HPWS que ante la sospecha del posible diagnóstico en este paciente, por su similitud a casos anteriormente vistos, realizó la interconsulta con el IHI, que acumula experiencia en estos enfermos, pues muchos cuadros de este tipo son interpretados como trombocitopenias secundarias a infecciones y en algunos, como manifestaciones de CID, y al no tener un manejo inmediato apropiado, el paciente puede fallecer. Aunque esta es una entidad poco frecuente, debe tenerse presente en neonatos con cuadros similares.
REFERENCIAS BIBLIOGRÁFICAS
1. Soult JA, García V, Sánchez MJ, Muñoz M, López JD, Tovaruela A. Síndrome de activación del macrófago: un reto diagnóstico. An Esp Pediatr 2002;56:165-7.
2. Farquhar J, Claireaux A. Familial haemophagocytic reticulosis. Arch Dis Child 1952;27:519 -5.
3. Kagi D, Odermatt B, Mak TW. Homeostatic regulation of CD8+ T cells by perforin. Eur J Immunol 1999;29:3262-72.
4. Risdall RJ, McKenna RW, Nesbit ME, Krivit W, Balfour HH, Simmons RL, et al. Virus associated hemophagocytic syndrome. A benign histiocytic proliferation distinct from malignant histiocytosis. Cancer 1979; 44:993-6.
5. Ningsanond V. Infection associated hemophagocytic syndrome: A report of 50 children. J Med Assoc Thai 2000;83:1141-9.
6. Henter J, Horne A, Arico M, Maarten R, Filipovich A, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48:124-31.
7. Janka GE, Schneider EM. Modern management of children with haemophagocytic lymphohistiocytosis. Br J Haematol 2004;124:4-14.
Recibido: 15 de marzo del 2010.
Aprobado: 2 de abril del 2010.
Dr. Juan Carlos Jaime Fagundo. Instituto de Hematología e Inmunología. Apartado 8070, Ciudad de La Habana, CP 10800, Cuba. Tel (537) 643 8695, 643 8268, Fax (537) 644 2334. e-mail: ihidir@hemato.sld.cu
Website: http://www.sld.cu/sitios/ihi


{kind=link}