










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter v.27 n.1 Ciudad de la Habana ene.-mar. 2011
ARTÍCULOS ORIGINALES
Policitemia Vera. Experiencias en el diagnóstico y tratamiento en el Instituto de Hematología e Inmunología
Polycythemia vera: Experiences in diagnosis and treatment in the Institute of Hematology and Immunology
Dra. Norma Fernández-Delgado, Ing. Teresa A. Fundora-Sarraf, Dra. Ivis Macías-Pérez
Instituto de Hematología e Inmunología. Ciudad de La Habana, Cuba.
RESUMEN
La policitemia Vera se encuentra entre las neoplasias mieloides según la clasificación más reciente de la Organización Mundial de la Salud (OMS). Sus criterios diagnósticos han tenido variaciones en los últimos años y en este trabajo se realiza un análisis de estos criterios, así como de la respuesta a los tratamientos empleados en 349 pacientes atendidos en el Instituto de Hematología e Inmunología en los últimos 40 años. Se sugiere, dada su factibilidad y eficacia, continuar utilizando por el momento la clasificación OMS del 2001, y de acuerdo con la experiencia acumulada, se propone el tratamiento con medicamentos de primera y segunda líneas según la edad y las características clínicas de cada enfermo.
Palabras clave: policitemia Vera, criterios diagnósticos, hidroxiurea, fósforo-32, flebotomía, interferón a.
ABSTRACT
The Polycythemia vera is located among the myeloid neoplasms according to the more recent classification of the HWO. Its diagnostic criteria have underwent variations in past years and in present paper authors made an analysis of such criteria, as well as of the response to treatments applied in 349 patients seen in the Institute of Hematology and Immunology during the past 40 years. Due to feasibility and effectiveness, for the moment, is has been suggested to carry on with the use of the classification of WHO of 2001, and according to the experience gained, it is proposed the treatment with first and second line drugs by age and the clinical features of each patient.
Key words: Polycythemia Vera, diagnostic criteria, hydroxyurea, phosphorus-32, phlebotomy, á-Interferon.
INTRODUCCIÓN
La policitemia Vera (PV) o enfermedad de Vaquez-Osler, es una dolencia hematológica caracterizada por proliferación clonal de los progenitores hematopoyéticos y que se expresa por un incremento absoluto de la masa eritrocitaria independiente de la eritropoyetina (Epo), leucocitosis, trombocitosis y esplenomegalia en más del 70 % de los casos. Otras de sus características son el prurito, la predisposición a trombosis, la fibrosis de la médula ósea y la transformación a leucemia aguda (LA) que puede ocurrir en un número reducido de enfermos.1,2
Recientemente, la Organización Mundial de la Salud (OMS) publicó cambios importantes en la clasificación de las neoplasias mieloides e incluyó a la PV entre las neoplasias mieloproliferativas.3
La PV aparece usualmente en individuos mayores de 60 años, con una incidencia mínima anual de 2,6 casos por 100 000 personas.2 Se incrementa con la edad, es rara en individuos menores de 40 años y también se ha comunicado eventualmente en niños.4,5
Los criterios para su diagnóstico han sufrido algunas transformaciones desde los emitidos por el Grupo Internacional de Estudios de la Policitemia Vera (GIEPV) en 1975,6 pero siempre se ha mantenido la demostración de la eritrocitosis clonal como un criterio mayor, y existen cambios en los criterios mayores y menores relacionados fundamentalmente con los avances en el campo del diagnóstico del laboratorio.7-9
Hace pocos años se descubrió que una mutación somática puntual de la tirosin cinasa JAK2, que consiste en la sustitución de la valina de la posición 617 por fenilalanina (JAK2V617F), está asociada con la PV y relacionada con su patogenia.2 Sin embargo, alrededor del 10 % de los pacientes con PV clásica no expresan esta mutación, que se puede expresar también en otras enfermedades mieloproliferativas crónicas como la mielofibrosis idiopática (MF) y la trombocitemia esencial,2,10-12 por lo que la sola expresión de esta mutación no debe bastar para el diagnóstico de PV. Posteriormente, en algunos pacientes con PV negativos para esta mutación, se describió una mutación similar en el exón 12 de la JAK2 asociada con eritrocitosis, pero sin la ocurrencia de panmielosis.13 Algunos autores han considerado que este evento genético tiene poca influencia en la clínica y el pronóstico de los pacientes con PV,10-11 pero a pesar de ello ha sido incluido entre los criterios diagnósticos mayores para la enfermedad.
El tratamiento de la PV está dirigido a disminuir las complicaciones, prevenir la transformación e incrementar la supervivencia; y con ese objetivo, medicamentos como la hidroxiurea (HU) o hidrocarbamida, el interferón a recombinante (IFN ar), el anagrelide y el Fósforo-32 (P32), han mostrado su efectividad en el control de la enfermedad. El pipobroman, derivado de la piperazina, que actúa como agente alquilante, también se ha empleado, particularmente en algunas regiones de Europa. Otros quimioterápicos como el clorambucil, la mostaza nitrogenada, el busulfan, la 6 tioguanina, también han sido usados.14-16
En este artículo se muestran las experiencias en el Instituto Hematología e Inmunología (IHI) en el diagnóstico y tratamiento de la PV en los últimos 40 años.
DIAGNÓSTICO
En los últimos años, la OMS publicó cambios importantes en los criterios diagnósticos de PV;3,7-9 los que se muestran en la tabla 1 comparados con los establecidos por el GIEPV.
Tabla 1. Criterios diagnósticos de policitemia Vera (PV)
| | Grupo Internacional de Estudios de la PV6 | | |
| Criterios mayores | |||
| A1 | Volumen globular: | Volumen globular > 25 % por encima del normal o Hb > 18,5 g/dL en el hombre y de >16,5 g/dL en la mujer | Hb > 18,5 g/dL en el hombre y de >16,5 g/dL en la mujer u otra evidencia de incremento absoluto del volumen globular |
| A2 | Saturación de O2 > 92% | Ausencia de causas secundarias | Presencia de la mutación JAK2 V617F u otra funcionalmente similar, como |
| A3 | Esplenomegalia palpable | Esplenomegalia palpable | - |
| A4 | - | Evidencia de clonalidad no Ph + ó BCR/ABL+ | - |
| A5 | - | Crecimiento espontáneo de colonias eritroides | - |
| Criterios menores | |||
| B1 | Plaquetas > 400 x 109/L | Plaquetas > 400 x 109/L | Biopsia de médula con hipercelularidad para la edad, crecimiento trilineal (panmielosis) con proliferación eritroide prominente, granulocítica y magacariocítica |
| B2 | Leucocitos > 12 x 109/L | Leucocitos > 12 x 109/L | Disminución de eritropoyetina sérica |
| B3 | Aumento de | Biopsia de médula ósea con características típicas de PV | Crecimiento espontáneo de colonias eritroides in vitro |
| B4 | Aumento de la vitamina B12 > 900pg/mL o UBBC > 2200 pg/ mL | Disminución de eritropoyetina sérica | - |
| Diagnóstico de PV | A1 + A2 + A3 | A1 + A2 + cualquier otro criterio AA1 + A2 + 2 criterios B | A1 + A2 + 1 criterio B |
En el IHI, durante más de 20 años, los pacientes con PV se clasificaron de acuerdo con los criterios del GIEPV y posteriormente se incluyó el estudio de la médula ósea (aspirado, cariotipo y biopsia) para el diagnóstico; estos últimos aparecen incluidos en los criterios de la OMS publicados en el 2001.17 En pocos casos fue posible realizar la cuantificación de Epo sérica y los cultivos de médula ósea.18 Como aún no se ha introducido en la institución el estudio molecular de la PV, se han continuado utilizando los criterios establecidos por la OMS en el 2001, ya que permiten establecer el diagnóstico en cualquier lugar con un mínimo de condiciones, siempre que se cumplan 2 de los primeros criterios mayores (A) más cualquier otro mayor; o los 2 primeros (A) con 2 menores (B). Los estudios ferrocinéticos también se han empleado en el diagnóstico y seguimiento de nuestros pacientes y reflejaron que el recambio de hierro plasmático y el tiempo medio de Fe-59 son los primeros parámetros en retornar a la normalidad después del tratamiento y los primeros en alterarse cuando se inicia el descontrol de la enfermedad. Ambos parámetros reflejan hiperactividad eritropoyética y pudieran considerarse como medidas indirectas de la eritropoyesis en el diagnóstico de la PV.19,20
TRATAMIENTO
El conocimiento acumulado de la historia natural de la PV ha permitido comprobar la progresión y severidad de la enfermedad cuando no se aplica tratamiento. La mitad de los enfermos no tratados fallecen por complicaciones trombóticas o hemorrágicas antes de los 18 meses posteriores al inicio de la sintomatología. 8,21,22
En el IHI se acumula una experiencia de más de 40 años en el tratamiento de la PV. En este acápite se analiza el efecto de diferentes tratamientos empleados. Un total de 349 pacientes con PV han sido tratados en este tiempo y para ello se han empleado fundamentalmente 4 modalidades terapéuticas: flebotomías, P32, HU e IFN ar.
Antes de expresar las variantes de tratamiento empleadas en nuestros pacientes, es oportuno realizar algunas definiciones relacionadas con elementos que se mencionan con posterioridad.
Riesgo trombótico:
Está presente en aquellos pacientes mayores de 60 años con antecedentes de eventos trombóticos, trombocitosis 600 x 109/L, o enfermedades concomitantes de riesgo como diabetes mellitus, hipertensión arterial, hipercolesterolemia y hábito de fumar.
Enfermedad progresiva:
Se identifica así cuando en cualquier momento de su evolución se encuentran uno o más de los siguientes resultados: incremento de leucocitos > 14 x 109/L, trombocitosis > 600 x109/L, esplenomegalia progresiva y descontrol del hematócrito (Hto) (aumento de más del 10 % del Hto en 3 meses o más de 6 flebotomías en un año). Presencia de síntomas B (pérdida de peso inexplicable, prurito intratable, sudaciones nocturnas, fiebre sin infección).
Control hematológico de la enfermedad:
Este se alcanza cuando se logran los siguientes resultados: Hto < 050, leucocitos < 12 x 109/L, plaquetas < 400 x 109/L, no más de 6 flebotomías en 1 año.
Flebotomías
La manifestación más evidente de la PV es el incremento de la masa de glóbulos rojos y por ende, del Hto. Las flebotomías han sido empleadas en el control de la eritrocitosis por años, pero es obvio que aunque es una medida eficaz en la reducción rápida del Hto, no tiene acción sobre otras manifestaciones como la leucocitosis, la trombocitosis y la esplenomegalia.9
Según el primer estudio aleatorizado del GIEPV,23 la mediana de supervivencia de los pacientes tratados con flebotomías era de 13,9 años, pero la mortalidad aumentaba en los primeros 4 años fundamentalmente por complicaciones trombóticas. Basados en los objetivos del tratamiento y las experiencias de este grupo, solo usamos la flebotomía como única opción terapéutica en aquellos pacientes sin riesgo trombótico y sin elementos de enfermedad progresiva.
La flebotomía es usada comúnmente como apoyo a otras alternativas terapéuticas cuando el Hto está por encima de 048. La cantidad a extraer oscila habitualmente entre 200 y 500 mL de acuerdo con la edad del paciente, y generalmente se realiza la reposición con solución salina fisiológica volumen a volumen, sobre todo en los pacientes mayores de 60 años.
La flebotomía se usó como única opción terapéutica en 85 pacientes de nuestra casuística, con respuesta del Hto en 47 (55,2 %); no obstante, solo 8 de los que respondieron inicialmente no requirieron posteriormente la utilización de otra opción de tratamiento.
Citorreducción
El Comité Británico de Estandarización en Hematología15,24 recomienda la citorreducción con HU e IFN como drogas de primera línea y el anagrelide, P32 y busulfan intermitente, como medicamentos de segunda línea, teniendo en cuenta alguna de las siguientes situaciones:
- Pobre tolerancia a las flebotomías.
- Esplenomegalia sintomática y progresiva.
- Trombocitosis.
- Cualquier otra evidencia de progresión de la enfermedad.
Hidroxiurea
A finales de los años 70 del pasado siglo, el GIEPV seleccionó la HU como el medicamento no mutagénico de elección para el tratamiento de la PV. La HU es un antimetabolito que actúa en la biosíntesis del ADN por inhibición de la enzima ribonucleótido reductasa y detiene el ciclo celular en fase de síntesis.25
A finales de la década de los 80, se realizó el primer reporte de este tratamiento en nuestro centro.26 Se obtuvo el 78 % de respuesta a los 3 meses de tratamiento. Resultados similares fueron reportados por otros autores27-29 y coinciden con nuestro análisis actual, donde de un total de 257 pacientes tratados con HU, en 194 (55,5 %) se usó como medicamento de primera línea con respuesta satisfactoria en el 79 % (n=153). La dosis inicial estuvo entre 15 y 30 mg/kg de peso corporal y se mantuvo hasta obtener un hematócrito < de 048. La dosis de mantenimiento fue de 15 mg/kg y la media de duración del tratamiento continuo de 2 años (rango 1 y 5 años). Cuando el Hto era ³ 050 se realizaron flebotomías hasta alcanzar un valor por debajo de esta cifra de Hto.
El seguimiento se realizó quincenalmente durante los primeros 3 meses y posteriormente fue mensual. Los efectos adversos encontrados en orden de frecuencia fueron: dispepsia, lesiones de piel, úlceras maleolares y anemia macrocítica. Ningún paciente que recibió la HU como única terapéutica desarrolló LA.
Interferón a recombinante
Reconocidos como inhibidores de la hematopoyesis, los interferones (a, b y g) ejercen un efecto inhibitorio directo sobre los progenitores pluripotentes.30 El IFN a ha sido usado en el tratamiento de la PV y es teóricamente la mejor opción para el tratamiento de esta enfermedad, pues no se ha reportado mutagenicidad.24
En nuestra serie, el IFN ar se ha utilizado en 38 pacientes, todos menores de 50 años, incluidas entre ellos 2 embarazadas con riesgo trombótico. En 36 casos (95 %) se usó como tratamiento de primera línea. Se obtuvo respuesta satisfactoria en el 52,7 % (n =19) y corresponde a los pacientes con trombocitosis > 600 x 109/L la mejor y más rápida respuesta. La media de tratamiento fue de 2 años. Doce pacientes (16,6 %) abandonaron el tratamiento antes de los 3 meses. Las causas fundamentales de esta conducta estuvieron relacionadas con la vía y frecuencia de la administración seguida de las reacciones adversas. El 82 % de los pacientes tratados con IFN ar presentó reacciones adversas de ligeras a moderadas, entre ellas: artralgias, mialgias, fiebre, caída del cabello y pérdida de peso. Las 3 primeras fueron disminuyendo progresivamente con las dosis subsiguientes. Para minimizar algunos de los efectos adversos se indicó duralgina una hora antes y una hora después de la administración del IFN ar.
Fósforo 32
Si se tiene en cuenta que un tratamiento es eficaz cuando aumenta la calidad de vida y la supervivencia del paciente, se observa que con la mielosupresión radioterápica con P32 se alargó el tiempo de supervivencia de los enfermos con PV por más de 10 años, con una excelente calidad de vida.21 Posteriormente, esto ha dado lugar al surgimiento de una interrogante sobre si su eficacia terapéutica incrementa el riesgo de LA. Este último es un problema de gran importancia porque la leucemia aguda secundaria es incurable en pacientes con PV. Por otra parte, varias series de pacientes tratados con P32 y seguidos por largo tiempo, han demostrado la eficacia de este radionúclido en limitar la panmielosis hiperplástica en la PV.15, 24,31-34
El P32 es un radioisótopo emisor de radiación beta (b) pura. Las radiaciones b emitidas producen su ionización en la vecindad inmediata de los átomos de fósforo depositados; este material ofrece la posibilidad de la irradiación local en enfermedades de la médula ósea. El isótopo inicialmente se concentra selectivamente en las células de la médula ósea activas mitóticamente y en unos pocos días es incorporado en el fosfato de calcio del hueso adyacente al endósteo, desde el cual se produce más irradiación de la médula. El P32 mantiene las características químicas del isótopo estable y sufre las mismas reacciones químicas en el organismo que el fósforo normal. Esto explica su toxicidad mínima y su metabolismo normal.14,16,31,32
La dosis de P32 recomendada por el GEPV es 2,3 mCi/ m2 de superficie corporal, con una dosis máxima de 5 mCi por dosis aplicada y de 15 mCi por año. Si no hay respuesta a la primera dosis, una segunda y tercera dosis pueden ser administradas con intervalos de 3 meses y solo con el 25 % de incremento en la dosis en cada ocasión.9,35
Por su efecto leucemogénico, el P32 debe ser reservado para pacientes mayores de 60 años. Siguiendo las recomendaciones de la EANM36 (European Association of Nuclear Medicine) su empleo fue considerado en:
1. Pacientes mayores de 60 años que no respondían a la terapéutica con flebotomías o tenían altos requerimientos de ellas (más de 6 por año) y no toleraban la HU.
2. Presencia de panmielosis rebelde a los tratamientos.
3. Trombocitosis > 600 x 109/L.
4. Como terapéutica de segunda línea en pacientes que no responden al tratamiento con HU e IFN ar.
5. Evidencia de enfermedad progresiva.
El seguimiento se realizó mensualmente con hemogramas en los primeros 3 meses posteriores a la administración del radiofármaco. Posteriormente, el seguimiento se realizó de acuerdo con la evolución del paciente, teniendo en cuenta si requería nuevas dosis. Se hizo estudio hematológico completo anual, por la posibilidad de desarrollo de una LA.
Un estudio que incluyó el análisis retrospectivo de todos los pacientes diagnosticados como PV,33 mostró 93 % de respuesta completa al tratamiento con P32. Hasta el momento, el P32 fue empleado en el 22,6 % (n= 77) de los pacientes con PV tratados en el IHI, el 98 % mayores de 60 años y cumplían al menos 2 de los criterios recomendados para la citorreducción. Las dosis del isótopo fueron las recomendadas por el GIEPV y se usó como tratamiento de primera línea en 31 pacientes. El porcentaje de remisión completa fue del 87 % (n=27) y otro 6 % (n =2) presentó remisión parcial.
La media de control hematológico mantenido postratamiento con P32 en nuestra serie fue de 28,6 meses, moderadamente superior al promedio reportado en la literatura.34 La supervivencia posterior a la administración del isótopo fue de 10,3 años y la supervivencia global fue de 17,2 años (rango entre 6 y 25 años).
El tratamiento de la PV con P32 permitió el control del recuento periférico de la sangre en los 2 meses posteriores a la administración del radiofármaco, así como el alivio de los síntomas y signos clínicos de la enfermedad y la disminución de la esplenomegalia en la mayor parte de nuestros pacientes.
Los efectos adversos al P32 se presentaron en el 6,4 % (n=5) de los pacientes y correspondieron con leucopenia y trombocitopenia moderadas, que se observaron después de las 4 semanas de tratamiento y que resolvieron espontáneamente.
Después del tratamiento con P32, 6 pacientes (7,8 %) desarrollaron una leucemia aguda mieloide (LMA). Al comparar este resultado con los que desarrollaron LMA y no habían recibido nunca el radioisótopo, la diferencia no fue significativa (p=0,09) (tabla 2).
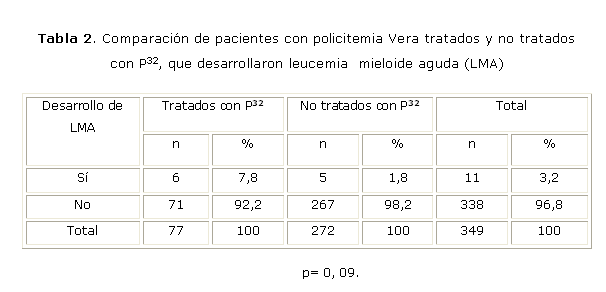
Los resultados de nuestro trabajo muestran una transformación a LA ligeramente menor y una eficacia superior a las de otras series publicadas. Además, apoyan el criterio de otros autores que afirman que la transformación leucémica es parte de la historia natural de la PV y que este riesgo puede incrementarse con los medicamentos que se emplean habitualmente en su tratamiento15,16,21,31. No obstante, sobre todo en pacientes mayores de 60 años, el beneficio puede exceder el riesgo potencial y el P32 puede ser usado en pacientes cuidadosamente seleccionados y con previo conocimiento del riesgo-beneficio que entraña este tratamiento.
De acuerdo con la experiencia acumulada, consideramos que el tratamiento con P32 es el método más conveniente para el enfermo añoso, que logra una buena calidad de vida y prolonga la supervivencia; además, no hay enfermedad de la radiación asociada con su uso y el paciente no tiene que ingerir medicamentos cuya dosis deba ser regulada constantemente. Otro aspecto importante es que las remisiones se producen en un período de tiempo relativamente corto después de su administración y pueden ser mantenidas por mucho tiempo sin requerir otra medicación ni atención médica frecuente.
OTROS TRATAMIENTOS
En nuestra serie, el busulfan se empleó en 5 pacientes, en 3 de ellos como tratamiento inicial a comienzos de la década de 1970. Otros 2 pacientes que no respondieron a la terapéutica empleada, fueron tratados posteriormente con este medicamento, sin respuesta satisfactoria; en uno de ellos se observó bicitopenia severa (leucopenia y trombocitopenia) sin respuesta eritrocitaria.
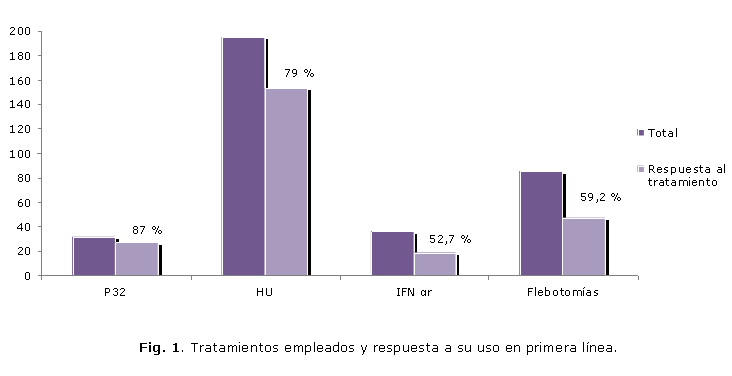
Un resumen de los tratamientos citorreductores empleados en nuestros pacientes con el porcentaje de respuesta a su empleo en primera línea, se muestra en la figura 1.
En los 349 pacientes, la media de seguimiento fue de 15 años (rango entre 1 y 22). De ellos, 13 pacientes tienen más de un año sin asistir a consulta al momento de redactar este trabajo.
El 12 % (42 pacientes) falleció y las causas de muerte están encabezadas por las complicaciones trombóticas, seguida de la LA y la MF (fig. 2). En el 72 % de los casos tratados con agentes citorreductores, la muerte ocurrió por encima de los 75 años, tiempo similar a la esperanza de vida de nuestra población, lo que significa que en estos pacientes se logró una supervivencia similar a la de la población normal.

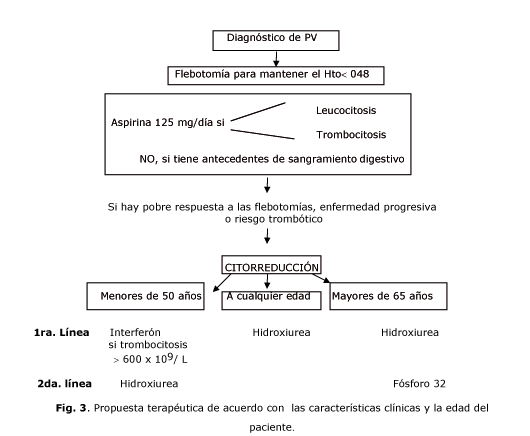
Una propuesta terapéutica de acuerdo con las características clínicas y la edad se muestra en la figura 3.
FUTURO
Algunos autores plantean que el tratamiento con IFN ar induce disminución de la expresión de la mutación JAK237 y que el pegylado induce remisiones hematológicas y moleculares completas en pacientes con PV.38,39 Además de estos beneficios, el IFN a 2a pegylado también podría ser una opción terapéutica más aceptada por los enfermos, pues al disminuir el número de inyecciones periódicas, probablemente reduciría el número de abandono del tratamiento.
La introducción del estudio molecular de la PV en el IHI, sin lugar a dudas, abrirá nuevas perspectivas diagnósticas y terapéuticas. Los inhibidores de la tirosina cinasa y especialmente los específicos de la JAK2, tienen un futuro promisorio en el tratamiento de la PV. El mesilato de imatinib ha mostrado inhibición del crecimiento autónomo de colonias eritroides in vitro y se ha usado en un pequeño número de casos con buenos resultados.20-22,37.
REFERENCIAS BIBLIOGRÁFICAS
1. Finazzi G, Barbui T. The treatment of polycythaemia vera: An update in the JAK2 era. Intern Emerg Med. 2007;2:13-8.
2. Tefferi A, Spivak JL. Polycythemia vera: Scientific advances and current practice. Semin Hematol. 2005;42:206-20.
3. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood. 2009;114:937-51.
4. Danish EH, Rasch CA, Harris JW. Polycythemia vera in childhood; a case report and review of the literature. Am J Hematol. 1980;9:421-8.
5. Fernández N, Fundora T, Silva R, Martínez L, García O, García M. Policitemia vera en niños: Reporte de 3 casos. Rev Cubana Hematol Inmunol Hemoter [serie en internet]. 2006; 22(3): Disponible en:
http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892006000300008&lng=es&nrm=iso&tlng=es
6. Turkington RC, Arnold EC, Percy MJ, Ranaghan LA, Cuthbert RJ, McMullin MF. Comparison of diagnostic criteria for polycythaemia vera. Hematology. 2007;12:123-30.
7. Michiels JJ, De Raeve H, Berneman Z, Van Bockstaele D, >Hebeda K, Lam K, et al. The 2001 World Health Organization and updated European clinical and pathological criteria for the diagnosis, classification, and staging of the Philadelphia chromosome-negative chronic myeloproliferative disorders. Semin Thromb Hemost. 2006;32:307-40.
8. Streiff MB, Smith B, Spivak L. The diagnosis and management of polycythemia vera in the era since the Polycythemia Vera Study Group: A survey of American Society of Hematology members practice patterns. Blood. 2002;99:1144-9.
9. Spivak JL, Silver RT. The revised World Health Organization diagnostic criteria for polycythemia vera essential thrombocitosis, and primary myelofibrosis: An alternative proposal. Blood. 2008;112:231-9.
10. Guglielmelli P, Barosi G, Pieri L, Antonioli E, Bosi A, Vannucchi AM. JAK2V617F mutational status and allele burden have little influence on clinical phenotype and prognosis in patients with post-polycythemia vera and post-essential thrombocythemia myelofibrosis. Haematologica. 2009;94:144-46.
11. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. editors. WHO Classifications of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon: IARC Press; 2008.
12. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood. 2009;114:937-51.
13. Williams DM, Kim AH, Rogers O, Spivak JL, Moliterno AR. Phenotipic variations and new mutations in JAK2V617F-negative polycythemia vera, erythrocytosis and idiophatic myelofibrosis. Exp Hematol. 2007;35:1641-6.
14. Fruchtman SM, Wasserman LR. Therapeutic recommendations for polycythemia vera. In: Wasserman LR, Berk PD, Berlin NI, editors. Polycythemia vera and the myeloproliferative disorders. Philadelphia: WB Saunders; 1995. p. 337-49.
15. McMullin MF. A review of the therapeutic agents used in the treatment of polycythaemia vera. Hematol Oncol. 2007;25:58-65.
16. Nilsson J, Westlin JE. Phosphorus-32 therapy in myeloproliferative disease. In: Ell PJ, Gambhir SS. Nuclear Medicine in clinical diagnosis and treatment. 3rd ed. London: Churchill Livingstone; 2004. p. 403-5.
17. Pierre R, Imbert M, Thiele J, Vardiman JW, Brunning RD, Flandren G. In Polycythaemia vera. In: Jaffi ES, Harris NL Stein H, Vardiman TW, editors. WHO Classification of Tumours: Tumours of haematopietic and lymphoid tissues. Lyon: IARC Press; 2001. p. 32-38.
18. García Y, Gudim VI, Callejas J. Determinación de eritropoyetina en plasma de pacientes con policitemia. Rev Cubana Hematol Inmunol Hemoter. 1985;1:56-61.
19. García Y, Callejas J, Hernández P. Use of ferrokinetic in the follow-up of patients with polycythaemia vera. Haematologica. 1988;1:41-6.
20. Fundora TA, Fernández N, Gramatges A, Lam RM. Valor del T½ del Fe-59 en el diagnóstico diferencial de la policitemia vera y la policitemia absoluta de causa secundaria. Rev Cubana Hematol Inmunol Hemoter [serie en internet]. 2003;19: Disponible en:
http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892003000100009&lng=es
21. Sánchez J, Román A, Nevado I. El tratamiento eficaz de una hemopatía incurable: la policitemia vera. Sangre. 1997;42:215-18.
22. Peterson P, Wasserman LR. The natural history of polycythemia vera. In: Wasserman LR, Berk PD, Berlin NI, editors. Polycythemia Vera and the myeloproliferative disorders. Philadelphia: WB Saunders; 1995. p. 14-21.
23. Berk PG, Goldberg JD, Donovan PB, Fruchtman SM, Berlin NF, Wasserman LR. Therapeutic recommendations in polycythemia vera based on Polycythemia Study Group protocols. Semin Hematol. 1986;23:132-43.
24. McMullin MF, Bareford D, Campbell P, Gree AR, Harrison C, Hunt B, et al. Guidelines for the diagnosis, investigation and management of polycythemia/erithrocytosis. Br J Hematol. 2005;130:174-95.
25. Finazzi G, Barbui T. How I treat patients with polycythemia vera. Blood. 2007;109:5104-11.
26. García Y. Tratamiento de la Policitemia vera con Hidroxiurea. Rev Cubana Hematol Inmunol Hemoter. 1992;8:94-99.
27. Donovan PB, Kaplan ME, Goldberg JD. Treatment of polycythemia vera with hydroxuyrea. Am J Hematol. 1984;17:329-34.
28. Kaplan ME, Mack K, Goldberg JD, Donovan PB, Berk PD, Wasserman LR. Longterm management of polycythemia vera with hydroxyurea. A progress report. Semin Hematol. 1986;23:161-71.
29. Najean Y, Rain JD. Treatment of polycythemia vera: The use of hydroxyurea and pipobroman in 292 patients under de age of 65 years. Blood. 1997;90:3370-7.
30. Putintseva E. Factores inhibidores de la hematopyesis. Presente y futuro. Sangre. 1996;41:459-53.
31. Parmentier C. Use and risk of phosphorus-32 in the treatment of polycythaemia vera. Euro J Nucl Med Mol Imaging. 2003;30:1413-7.
32. Roberts BE, Smith AH. Use of radioactive phosphorus in haematology. Blood Rev. 1997;11:146-53.
33. Fernández N, Fundora T, Milanés MT. Fósforo 32: experiencia de 30 años en la policitemia vera. Rev Cubana Hematol Inmunol Hemoter [serie en internet]. 2003;19(2-3): Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892003000200013&lng=es
34. Balan KK, Critchley M. Outcome of 259 patient with primary proliferative polycythaemia (PPP) and idiopathic thrombocythaemia (IT) treated in a regional nuclear medicine department with phosphorus-32a 15 year review. Br J Radiol. 1997;70:1169-73.
35. Berk PD, Wasserman LR, Fruchtman SM, Goldberg TJ. Treatment of polycythemia vera, a summary of clinical trends conduced by the polycythemia vera study sub-group. In: Wasserman LR, Berk PD, Berlin NI, editors. Polycythemia Vera and the myeloproliferative disorders. Philadelphia: WB Saunders; 1995. p. 166-194.
36. Tennvall J, Brant B. EANM procedure guidelines for 32P phosphate treatment of myeloproliferative diseases. Eur J Nucl Med Mol Imaging. 2007;34:1324-27.
37. Jones AV, Silver RT, Waghorn K, Curtis C, Kreil S, Zoi K, et al. Minimal molecular response in polycythemia vera patients treated with imatinib or interferon alpha. Blood. 2006;107:3339-41.
38. Kiladjian JJ, Cassinat B, Turlure P, Cambier N, Murielle R, Bellucci M, et al. High molecular response rate of polycythemia vera patients treated with pegylated interferon a- 2a. Blood. 2006;108:2037-40.
39. Kiladjian JJ, Cassinat B, Chevret S, Turlure P, Cambier N, Murielle R, et al. Pegylated interferon a- 2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood. 2008;112:3065-72.
Recibido: 15 de septiembre del 2010.
Aprobado: 1 de octubre del 2010.
Dra. Norma Fernández-Delgado. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800. Ciudad de La Habana, Cuba. Tel (537) 643 8695, 8268, Fax (537) 644 2334. correo electrónico: ihidir@hemato.sld.cu; ihi@infomed.sld.cu
Website: http://www.sld.cu/sitios/ihi/index.php

