










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión impresa ISSN 0864-0289
Rev Cubana Hematol Inmunol Hemoter vol.27 no.4 Ciudad de la Habana oct.-dic. 2011
MINI-REVISIÓN
El síndrome de las plaquetas pegajosas y su diagnóstico en el laboratorio
The sticky platelet syndrome and its diagnosis in the laboratory
MSc. Loreta Rodríguez Pérez, Dra. Dunia Castillo González
Instituto de Hematología e Inmunología. La Habana, Cuba.
RESUMEN
El síndrome de las plaquetas pegajosas (SPP) es un trastorno plaquetario autosómico dominante considerado como una de las causas más frecuentes de eventos trombóticos, tanto arteriales como venosos. Este síndrome supone trastornos en la agregación de las plaquetas caracterizados por su incremento anormal. El mecanismo patogénico no se conoce; su existencia puede determinarse con las pruebas de agregación y adhesión plaquetarias. Tampoco se conoce su prevalencia, pero hay datos que sugieren que es frecuente. Algunos investigadores plantean que es responsable del 23 % de las trombosis arteriales inexplicables y del 14 % de las venosas, en las que no es posible identificar una causa. Se presenta una breve revisión acerca de la información disponible sobre el síndrome de las plaquetas pegajosas y su trascendencia en la salud. En Cuba existen pocos reportes de la evaluación clínica de pacientes con esta afección, por lo que resulta necesario realizar estudios más profundos para establecer la magnitud de la enfermedad.
Palabras clave: Síndrome de las plaquetas pegajosas, trombosis, pruebas de hiperagregación y adhesión plaquetarias.
ABSTRACT
The sticky platelet syndrome (SPS) is an autosomal dominant platelet disorder considered as one of the most common causes of thrombotic events, both arterial and venous. This syndrome involves disturbances in platelet aggregation characterized by abnormal increase. The pathogenic mechanism is not known. Its existence can be determined by platelet aggregation and platelet adhesion tests. Its prevalence is also unknown, but evidence suggests that it is common. Some researchers argue that it is responsible for 23 % of unexplained arterial thrombosis and 14 % of the vein thrombosis, in which is not possible to identify a cause. Here a brief review on the available information on the sticky platelet syndrome and its importance in health is shown. In Cuba there are few reports of the clinical evaluation of patients with this condition, so further study to determine the extent of the disease is needed.
Key words: Sticky platelet syndrome, thrombosis, hyper aggregation and platelet adhesion tests.
INTRODUCCIÓN
El síndrome de las plaquetas pegajosas (SPP) es un trastorno plaquetario autosómico dominante considerado como una de las causas más frecuentes de eventos trombóticos, tanto arteriales como venosos, a pesar de ser para muchos una enfermedad desconocida.
Desde hace algún tiempo se le conoce como la segunda causa de los trastornos hereditarios relacionados con los problemas trombóticos, después de la resistencia a la proteína C activada, y parece corresponder con la principal causa de trombosis arteriales. Se cree que el defecto específico puede estar localizado en los receptores de la superficie plaquetaria y está caracterizado por un incremento anormal de la agregación de las plaquetas con difosfato de adenosina (ADP), epinefrina o ambas. Este síndrome constituye un fenómeno de hipercoagulación que cada vez se asocia más con accidentes tromboembólicos.1-3
ANTECEDENTES
En 1983, Holliday y otros, describieron por primera vez un síndrome relacionado con infarto cerebral en adultos jóvenes y lo nombraron "síndrome de la plaqueta pegajosa", pero su prevalencia no fue motivo de reconocimiento en la literatura médica hasta 1985.2,3
En 1986, Mammen y otros, publicaron el estudio de 41 pacientes adultos con dolor precordial y arterias coronarias angiográficamente normales, en los que encontraron hiperagregación plaquetaria en las pruebas realizadas con los agonistas ADP y epinefrina.4 Este estudio confirmó la importancia clínica del SPP. Aunque su causa no se conoce, su existencia puede definirse con las pruebas de agregación plaquetaria, para lo cual resulta importante descartar que se trate de un hallazgo de laboratorio.
En 1995 se publicó un nuevo estudio que incluyó más de 200 pacientes jóvenes entre 5 y 45 años y sus familiares, que presentaban trombosis, principalmente arteriales y venosas en varios casos, sin factores asociados. Todos mostraban hiperagregación con ADP, epinefrina o ambos.5
En un estudio realizado en 153 pacientes adultos con cuadros trombóticos se encontró que del total de trombosis venosas, el 14 % correspondió al SPP, y el 23 % a los eventos arteriales.6
CARACTERÍSTICAS CLÍNICAS
En la población adulta, el SPP se asocia con fenómenos anginosos, infarto agudo del miocardio, isquemia cerebral transitoria, cuadros de apoplejía y trombosis retiniana, trombosis de arterias periféricas y trombosis venosas frecuentemente recurrentes aun bajo tratamiento con anticoagulantes orales. La mayoría de los eventos son de tipo arterial.3
Por otra parte, las gestantes tienden a presentar síndrome de muerte fetal intrauterina. Además, se ha considerado que las situaciones de estrés emocional pueden activar mecanismos de hiperagregación plaquetaria.7,8
Algunos autores han relacionado la agregación plaquetaria con los ciclos circadianos y han encontrado un predominio de aparición en las horas de la mañana. También han descrito que este síndrome se puede asociar con otras anormalidades congénitas de los mecanismos de anticoagulación, como resistencia a la proteína C activada y deficiencias de proteína S.9
Se han descrito muchas enfermedades que cursan con hiperagregabilidad plaquetaria como: diabetes mellitus, síndrome nefrótico, fibrosis quística y anorexia nerviosa, entre otras. En estos casos se ha encontrado que los niveles de factor 4 plaquetario (PF4), b tromboglobulina y tromboxano A2, son elevados, lo que sugiere la existencia de mecanismos intrínsecos, probablemente secundarios, de activación plaquetaria en la circulación. También es importante tener en cuenta que los defectos congénitos de la lipooxigenasa, generan trombosis sin elevación del FP4 ni de la b tromboglobulina.10
DIAGNÓSTICO DE LABORATORIO
La mayoría de los autores fundamentan el diagnóstico en la demostración de las hipersensibilidades al ADP y la epinefrina mediante pruebas de agregometría.8 Sin embargo, se conoce que estas pruebas se han diseñado especialmente para la demostración de los defectos en la agregación y no para la respuesta plaquetaria aumentada.11
Uno de los sistemas para el estudio de las plaquetas pegajosas es el equipo PFA-100, considerado como uno de los más sensibles, sin restarle mérito a los ensayos de agregometría que son en general los más utilizados.11
Las hipersensibilidades se inducen con 3 concentraciones diferentes para cada agente agregante (tabla), y la agregación plaquetaria se mide en el agregómetro, donde se registran los cambios en la densidad óptica, manteniendo una temperatura de 37 °C y agitación a velocidad constante.
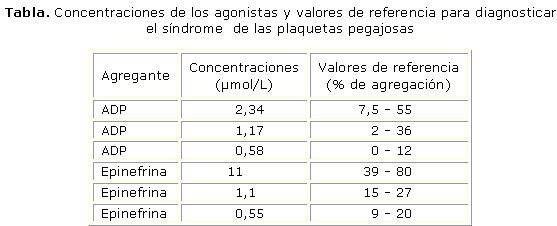
La agregación se expresa en porcentaje de transmisión de la luz: 100 % es agregación completa y 0 % ausencia de agregación.
En los casos normales, al diluir el ADP y la epinefrina, las curvas muestran una marcada disminución de la agregación, mientras que en los pacientes que presentan el SPP, las curvas de agregación permanecen elevadas.12
CRITERIOS DIAGNÓSTICOS
Los tipos del SPP se definen de acuerdo con los resultados obtenidos en la agregación:
- Tipo I: hiperagregación con epinefrina y con ADP.
- Tipo II: hiperagregación solo con epinefrina.
- Tipo III: hiperagregación solo con ADP.
En pacientes con antecedentes de trombosis, el diagnóstico se puede sospechar cuando existe hiperagregación con un solo reactivo y a una sola concentración. Esta sospecha se concreta cuando se repite la prueba y persiste la misma alteración.
La existencia de este síndrome se puede sospechar en aquellos pacientes con antecedentes de trombosis, en que hay hiperagregación con un solo reactivo y a una sola concentración. El diagnóstico se confirma cuando se repite la prueba y persiste la alteración.
Los datos que confirman el diagnóstico son la existencia de antecedentes de trombosis con hiperagregación plaquetaria con:
a) Dos concentraciones y 2 reactivos diferentes.
b) Una concentración con 2 reactivos diferentes.
c) Una sola concentración y 1 reactivo en 2 ocasiones diferentes.8
INDICACIONES PARA EL ESTUDIO
Se debe destacar que en todos los pacientes con SPP, las pruebas de agregación con colágeno, ácido araquidónico y ristocetina son normales.12
En los cuadros agudos de trombosis arteriales o venosas se han encontrado fenómenos de hiperagregabilidad que podrían ser consecuencia de activación del sistema hemostático. Por esta razón, las pruebas mencionadas se deben realizar o repetir de 4 a 9 meses después del evento trombótico.12
La investigación del SPP debe realizarse sin que el paciente haya ingerido, en los últimos 15 días, fármacos que inhiban la agregación plaquetaria, ya que de lo contrario, pueden obtenerse resultados falsos negativos.13
Para corroborar el diagnóstico, las pruebas se deben realizar en más de una oportunidad (3 veces como mínimo), con resultados similares.13
Los hallazgos de laboratorio deben desaparecer durante el tratamiento antiagregante y reaparecer cuando se repiten las pruebas al suspender el medicamento.7
Se debe investigar la existencia de un patrón familiar mediante las pruebas realizadas a padres, hermanos y otros familiares.7
Si las pruebas se han realizado durante el evento agudo, se deben repetir después de 4 y hasta 9 meses, con suspensión del antiagregante 15 días previos al estudio.13
Se deben realizar todas las demás pruebas de hipercoagulación, por lo menos las más frecuentemente descritas.7
TRATAMIENTO
La hiperagregación plaquetaria que caracteriza a estos pacientes se revierte habitualmente con el empleo de inhibidores de la agregación plaquetaria. Se recomienda el ácido acetilsalicílico (ASA) y existen informes de los resultados de este tratamiento en pacientes con SPP.7
El ASA inhibe la agregación plaquetaria por la inhibición selectiva de la ciclooxigenasa plaquetaria, lo que disminuye la producción del tromboxano A2 (potente vasoconstrictor y estimulador de la agregación plaquetaria) y modifica el equilibrio entre estas 2 sustancias antagónicas.
Una dosis de 80-100 mg/día de ASA es suficiente para reducir el riesgo de trombosis y lograr la normalización del patrón hiperagregante en la mayoría de los casos. En otros pacientes es necesario aumentar la dosis hasta 325 mg/día para lograr la normalización. Si no se observa mejoría, es necesaria entonces la utilización del clorhidrato de ticlopidina en dosis de 250-500 mg/día.1
Cuando se suspende el tratamiento, aparecen nuevamente las alteraciones en las pruebas de laboratorio y aumenta el riesgo de evento vasooclusivo.1
Debe destacarse que los resultados alcanzados en las investigaciones muestran cada vez más que el SPP presenta cierta tendencia hacia eventos trombóticos, tanto arteriales como venosos, y es considerado por muchos investigadores una enfermedad aún desconocida.8
A pesar de la escasa descripción de este síndrome, se han reportado fundamentalmente estudios en adultos con antecedentes de enfermedad tromboembólica venosa, infarto agudo del miocardio y enfermedad cerebrovascular. Sin embargo, su incidencia en la población femenina en estado de gestación ha sido poco estudiada, por lo que resulta importante tenerlo en cuenta para el diagnóstico diferencial de las trombofilias en pacientes embarazadas con antecedentes de pérdidas gestacionales y de trombosis venosa profunda.
Por otra parte, en Cuba no existen reportes de la evaluación clínica de pacientes con esta afección, por lo que resulta necesario realizar estudios más profundos para establecer la magnitud de la enfermedad en nuestra población.
REFERENCIAS BIBLIOGRÁFICAS
1. Ruiz Argüelles G, Ruiz Delgado G, López Martínez B. El síndrome de las "plaquetas pegajosas": una causa frecuente pero ignorada de trombofilia. Rev Invest Clin. 2002 Sep-Oct;54(5):394-6.
2. Holliday PL, Mammen EF, Gilroy J, Buday J, Barnhart M. Sticky platelet syndrome and cerebral infarction in young adults. 9th International Join Conference on Stroke and Cerebral Circulation, Phoenix AZ. 1983 (Abstracts).
3. Bick RL. Sticky platelet syndrome: A common cause of unexplained arterial and venous thrombosis. Clin Appl Thromb Hemost. 1998 Apr;4(2):77-81. DOI: 10.1177/107602969800400201
4. Mammen EF, Rubenfire M, Blevins RD, Barnhart M, Housholder S, Selik N. Platelet hyperaggregability in patients with chest pain and angiographically normal coronary arteries. Am J Cardiol. 1986 Mar 1;57(8):657-60.
5. _____. Ten years' experience with the "sticky platelet syndrome" Clin Appl Thrombosis/ Hemostasis. 1995 Jan 1;(1):66-72.
6. Bick RL, Kaplan H. Syndromes of thrombosis and hypercoagulability: Congenital and acquired thrombophilias. Clin Appl Thrombosis/Hemostasis. 1998 Jan;4(1):25-50. DOI: 10.1177/107602969800400106
7. Velásquez, Alvinzy II, Carmona V, Ramos G. El síndrome de la plaqueta pegajosa: serie de casos en gestantes en el Hospital Militar Central. Rev Colomb Obstet Gginecol. 2004 Sep;55(3):232-9.
8. Levine SP, Towel BL, Suárez AM. Platelet activation and secretion associated with emotional stress. Circulation. 1985 Jun;71(6):1129-34.
9. Chaturvedi S, Dzieczkowski JS. Protein S deficiency, activated protein C resistance and sticky platelet syndrome in a young woman with bilateral strokes. Cerebrovasc Dis. 1999;9:127-30.
10. Sinzinger H, Kaliman J, O'Grady J. Platelet lypoxigenase defect (Wien Penzing defect) in two patients with myocardial infarction. Am J Hematol 1991 Mar;36(3)202-5. DOI: 10.1002/ajh.2830360308
11. Mammen EF. Sticky platelet syndrome. Semin Thromb Hemost. 1999;25(4):361-5. DOI: 10.1055/s-2007-994939
12. Parra Ortega I, Estrada Gómez RA, Ruiz Argüelles GJ. Trombofilia multifactorial en México: síndrome de las plaquetas pegajosas. Medicina Universitaria. 2006 Jul;8(32):143-7.
13. _____. Síndrome de las plaquetas pegajosas, la condición de trombofilia heredada más frecuente en pacientes mexicanos. Medicina Universitaria. 2007 Ene;9(34):20-3.
Recibido: 7 de junio de 2011.
Aprobado: 25 de junio de 2011.
MSc. Loreta Rodríguez Pérez. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800. La Habana, Cuba. Tel (537) 643 8695, 8268, Fax (537) 644 2334. Correo electrónico: ihidir@hemato.sld.cu
Website: http://www.sld.cu/sitios/ihi

