










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión impresa ISSN 0864-0289
Rev Cubana Hematol Inmunol Hemoter vol.28 no.1 Ciudad de la Habana ene.-mar. 2012
ARTÍCULO DE REVISIÓN
Urgencias en Hematología: alteraciones metabólicas y leucocitarias
Emergencies in Hematology: metabolic and leukocyte alterations
Dr. Juan Carlos Jaime-Fagundo,I Dr. Alberto Arencibia-Núñez,I Dra. Adys Gutiérrez-Díaz,I Dr. Luis Ramón-Rodríguez,I Dra. Carmen Díaz-Durán,II Dr. Jesús Serrano-Mirabal,I Dra. Sandra Sarduy-Sáez,I Dra. Valia Pavón-MoránI
I Instituto de Hematología e Inmunología. La Habana, Cuba.
II Policlínico Docente "La Rampa". La Habana, Cuba.
RESUMEN
Las urgencias que pueden surgir durante la evolución de las enfermedades hematológicas son diversas, tanto por el comportamiento de la enfermedad de base como por el momento en que se presentan. Pueden ser la primera manifestación de la enfermedad o aparecer en el curso de su evolución, y sus secuelas pueden ser menores si se diagnostican y tratan adecuadamente. Se hace una revisión de algunas de las alteraciones metabólicas y leucocitarias más frecuentes que se pueden presentar en el paciente oncohematológico.
Palabras clave: urgencias hematológicas, neutropenia, leucocitosis, hipercalcemia, hiperviscosidad.
ABSTRACT
Emergencies which may appear during the evolution of hemopathies are diverse due to their basic behavior as well as according to the moment the disease comes into view. They may be the first sign of the disease or could appear during the course of its evolution. Consequences might be milder if diagnosed and treated properly. A revision of some of the most frequent metabolic and leukocyte alterations that could arise in the patient with an oncohematological disease is shown.
Key words: hematological emergencies, neutropenia, leukocytosis.
INTRODUCCIÓN
Los avances logrados en el tratamiento del paciente oncohematológico obligan a adoptar una actitud activa cuando aparecen complicaciones debidas a la evolución de la enfermedad o a los tratamientos administrados, con el objetivo de detectarlas y tratarlas sin demora. De esta forma, se consigue reducir al mínimo su repercusión y con ello los daños que pudieran emanar, con una mejoría de la calidad de vida de estos pacientes.
Las urgencias que pueden surgir durante la evolución de las enfermedades oncohematológicas son diversas, tanto por el comportamiento de la enfermedad de base, como por el momento en que se presentan. Pueden ser la primera manifestación de la enfermedad o aparecer en el curso de su evolución. Los adelantos logrados en los tratamientos de apoyo mediante el uso de antibióticos potentes, hemocomponentes y la administración de factores estimuladores de colonias, han dado lugar a un aumento en la supervivencia de los pacientes con cáncer y otras enfermedades hematológicas.
El tratamiento agresivo de una urgencia hematológica está indicado en muchas ocasiones, aún cuando no se haya establecido un diagnóstico exacto de la enfermedad de base. En el caso de la urgencia oncohematológica, cuando es consecuencia directa del proceso maligno, se debe tratar la neoplasia subyacente siempre que se disponga de un tratamiento efectivo para esta. El tratamiento debe iniciarse rápidamente para evitar complicaciones e incapacidad permanente. En este trabajo se revisan algunas de las complicaciones metabólicas y leucocitarias que se presentan frecuentemente en los pacientes hematológicos.
HIPERCALCEMIA
Definición
Trastorno que se produce cuando la movilización del calcio óseo supera la capacidad de eliminación renal. El aumento de calcio puede deberse a la destrucción ósea o a la producción por el tumor de sustancias que estimulan la actividad osteoclástica.1 Es una complicación metabólica muy frecuente en oncología y en el caso de los pacientes hematológicos, se presenta con mayor frecuencia en el mieloma múltiple (MM) y en algunos linfomas. En los primeros ocurre entre el 30 y 40 % de los casos y se encuentra asociada con infiltración importante de la médula ósea por células plasmáticas, lo que puede aparecer al inicio de la enfermedad o como elemento clínico que anuncia su recaída.1
A pesar de su frecuencia, frecuentemente no se diagnostica por falta de sospecha clínica al presentar síntomas que se atribuyen a otras causas en el contexto de la enfermedad, lo que impide su adecuado tratamiento.1,2
Nos referiremos a la hipercalcemia producida en los pacientes que sufren de MM, por ser la causa más frecuente en los pacientes hematológicos.
Etiopatogenia
Se han descrito 2 mecanismos o tipos de hipercalcemia inducida por el cáncer: la hipercalcemia osteolítica y la tumoral. Ambos mecanismos pueden actuar simultáneamente en algunos pacientes y se han encontrado niveles altos de calcitriol en pacientes con enfermedad de Hodgkin (EH), linfomas no hodgkinianos (LNH) y MM, aunque el primer mecanismo no es el más frecuente en las entidades hematológicas.1
En el MM, la hipercalcemia es debida a la amplia destrucción ósea inducida por el tumor. La actividad osteoclástica incrementada y la resorción ósea son producidas por potentes citocinas secretadas por las células mielomatosas, como las interleucinas 1 y 6, el activador del receptor nuclear del factor-κB, ligando RANKL, la proteína inflamatoria macrofágica [MIP]-1a, y el factor de necrosis tumoral (FNT).3
Hay factores que actúan como coadyuvantes, como la inmovilización del enfermo causada por la debilidad y la letargia asociadas con la hipercalcemia, que favorece la resorción ósea.3,4 También la anorexia, los vómitos y la poliuria (por calciuresis) ocasionan deshidratación con disminución del flujo renal y el filtrado glomerular, lo que ocasiona una mayor reabsorción de calcio.2 Esta resorción lleva a la salida de calcio al líquido extracelular que sobrepasa la capacidad renal de resorción y produce un exceso de calcio en la circulación, por lo que se ha clasificado la hipercalcemia en ligera, moderada y severa.
Clasificación de la hipercalcemia según calcio sérico:
- Ligera: calcio sérico entre 2,6 y 3 mmol/L.
- Moderada: calcio sérico entre 3 y 3,75 mmol/L.
- Grave: calcio sérico mayor que 3,75 mmol/L.
Los valores normales de calcio son: 8,5-10,2 mg/dL. (Para convertir de mg/dL a mmol/L, el factor de conversión es X 0,25.)
Los síntomas de la hiper o hipocalcemia, se deben a anormalidades en la fracción ionizada del calcio plasmático y son las variaciones del calcio iónico las verdaderamente importantes. Sin embargo, muy raras veces los laboratorios analizan de manera rutinaria los niveles de calcio ionizado.
El calcio plasmático total se usa para inferir la fracción del calcio ionizada, y usualmente es bastante preciso, excepto en los casos donde se presenta hipoalbuminemia. Debido a que esto no es raro en los pacientes con cáncer, se hace necesario "corregir" la concentración total de calcio plasmático, por aquel porcentaje de calcio que se hubiese podido medir, si los niveles de albúmina hubiesen estado dentro de los parámetros normales.5 Los cálculos son los siguientes:
Concentración total de calcio, corregida para los niveles de albúmina:
[(albúmina normal-albúmina del paciente) x 0,8] + calcio del paciente
(Valores normales de albúmina: 3,1-4,3 g/dL ó 31-43 g/L; factor de conversión X10)
Manifestaciones clínicas de la hipercalcemia
- Generales: deshidratación, pérdida de peso, prurito y astenia.
- Neurológicas: somnolencia ligera, cefalea, debilidad progresiva, depresión, letargia, ataxia, estupor y coma.
- Gastrointestinales: constipación, polidipsia, náuseas, vómitos, anorexia, úlcera péptica, y dolor abdominal.
- Músculo esquelético: debilidad, dolor óseo, y cansancio.
- Genitourinarios: polidipsia, poliuria, nefrolitiasis e insuficiencia renal crónica.
- Cardíacos: bradicardia, bloqueos, arritmias, asistolia, alargamiento del intervalo PR, acortamiento del intervalo QT, ensanchamiento del complejo QRS, alteraciones en el ST-T (ST acortado, T ancha), aumento de sensibilidad a los digitálicos.
Guía rápida de tratamiento inmediato
- Si calcemia < 12 mg/dL y el paciente está asintomático solo se administra hidratación amplia.
- Si calcemia entre 12-14 mg/dL y no hay síntomas, añadir furosemida al tratamiento. Si se presentan síntomas, entonces se pueden añadir bifosfonatos.
- Si calcemia > 14 mg/dL administrar hidratación, furosemida, calcitonina o mitramicina y bifosfonatos, y se evalúa el uso de corticoides si están indicados.
En situaciones de insuficiencia renal o si fracasan las anteriores medidas, considerar la hemodiálisis.
Tratamiento de la hipercalcemia
- Hidratación con solución salina 0,9 % (excepto que exista una contraindicación), como mínimo 2 000 mL/24 horas.
- Corticosteroides: disminuyen la resorción ósea y la absorción digestiva de calcio. Puede utilizarse prednisona (40-60 mg/m2/día, oral) o dexametasona (6-9 mg/m2/día, intravenosa) o metilprednisolona (30-50 mg/m2/día, intravenosa). Los esteroides deben reducirse y suspenderse lo antes posible.
- Iniciar lo antes posible la quimioterapia específica.
- Indicación de bifosfonatos en pacientes con función renal normal.
Si las medidas anteriores fracasan, se valora la indicación de la mitramicina o calcitonina.
- Calcitonina: aumenta la excreción renal de calcio y disminuye la resorción ósea. Dosis de 4-8 UI/kg cada 12 horas, por vía subcutánea o intramuscular. Tiene acción rápida (pico en 2 a 4 horas), pero débil. Tras 2 o 3 dosis se desarrolla taquifilaxia, que la convierte en ineficaz.
- Mitramicina: esta droga ejerce su efecto citotóxico directo sobre los osteoclastos en dosis de 15-25 µg/kg/día, por vía intravenosa, en 500 mL de solución salina a pasar en 4 a 6 horas (dosis total 1,5-2 mg). Sus efectos secundarios (insuficiencia renal, trombopenia y daño hepático) limitan su uso. La máxima acción se alcanza a las 12-24 horas.
- Diuréticos de asa: furosemida en dosis de 10 a 20 mg cada 6 horas, por vía intravenosa. Evitar el uso de tiazidas y con la precaución de haber rehidratado correctamente al enfermo.
- Disminuir el aporte de calcio y vitamina D.
- Si hay oligoanuria, aplicar métodos de diálisis con líquidos sin calcio.
Los bifosfonatos son análogos de los pirofosfatos inorgánicos en formas estables y sintéticas con gran afinidad por el calcio. Los disponibles incluyen los de primera generación (etidronato y clodronato) que no contienen nitrógeno y son metabolizados a análogos citotóxicos ATP y producen la muerte de los osteoclastos.6 Los más recientes contienen nitrógeno, dentro de los que se encuentran el pamidronato, ibandronato y el ácido zoledrónico (Zometa), los que se unen con la enzima pirofosfato farnesil sintetasa y llevan a la inhibición de una serie de proteínas como la GTPasa, Ras, Rac, y Rho, que tienen una acción clave en la regulación de la función osteoclástica y en los eventos de resorción ósea.7,8
- Pamidronato: dosis de 60 a 90 mg por vía intravenosa en un período de 2 a 24 horas y continuar con 3 dosis de 60 mg cada 2 semanas.
- Ácido zoledrónico (Zometa): la dosis recomendada en la prevención de eventos relacionados con el esqueleto en pacientes con neoplasias avanzadas con afectación ósea es de 4 mg. El concentrado debe ser diluido con 100 mL de cloruro sódico al 0,9 % o solución de glucosa al 5 % y administrado como perfusión intravenosa durante 15 min, como mínimo cada 3 o 4 semanas. Deberá administrarse a los pacientes diariamente un suplemento oral de calcio de 500 mg y 400 UI de vitamina D. Los pacientes deben mantenerse bien hidratados antes y después de la administración de este medicamento.8
Cuando se inicia el tratamiento con Zometa en pacientes con MM, se deberá determinar la creatinina sérica y el aclaramiento de creatinina (CrCl). El CrCl se calcula a partir de la creatinina sérica utilizando la fórmula de Cockcroft-Gault.9,10 No se recomienda Zometa en los pacientes que presenten insuficiencia renal grave, definida como CrCl<30 mL/min, antes del inicio del tratamiento. En pacientes con metástasis óseas y MM que presentan insuficiencia renal de leve a moderada, definida como CrCl 30-60 mL/min, antes del inicio de tratamiento, se recomienda las dosis de Zometa que se muestran en la tabla 1.6

SÍNDROME DE LISIS TUMORAL
Definición
Conjunto de manifestaciones clínico humorales, resultantes de la liberación abrupta al torrente sanguíneo de componentes intracelulares que producen alteraciones metabólicas graves. Esta urgencia oncológica se caracteriza por hiperuricemia, hiperfosfatemia e hiperpotasemia, así como hipocalcemia e insuficiencia renal aguda (IRA).11
Etiopatogenia
El síndrome de lisis tumoral (SLT) puede ocurrir por la ruptura espontánea de las células tumorales, o más comúnmente secundario a la quimioterapia o radioterapia citotóxicas. Una vez establecido acarrea complicaciones graves, por lo que la prevención constituye la piedra angular de su tratamiento. Entre el 15 y el 25 % de los pacientes con leucemias agudas o LNH avanzado (sobre todo de tipo Burkitt) presentan SLT al inicio de la quimioterapia citorreductora.11,12
Para que un tumor pueda causar un SLT debe cumplir 2 condiciones: tener una fracción de crecimiento alta y una elevada tasa de síntesis de purinas.
La fracción de crecimiento (FC) de un tumor es la resultante del número de células que se encuentran en el ciclo celular entre la cantidad total de células del tumor. Desde el punto de vista clínico, se consideran tumores con elevada FC aquellos cuyo porcentaje es mayor que el 30 %. Actualmente la medición se efectúa por inmunohistoquímica, calculando el porcentaje de positividad para el marcador Ki 67, una proteína que se expresa en el núcleo durante la proliferación celular.
En la siguiente lista se resumen algunos de los tumores hematológicos con mayor FC:
1. Linfomas no hodgkinianos tipo Burkitt = 100 %.
2. Linfomas no hodgkinianos de alto grado = 60-80 %.
3. Leucemia linfoide aguda = 50-75 %.
4. Leucemia mieloide aguda = 70 %.
5. Linfomas no hodgkinianos de grado intermedio = 40 %.
6. Leucemia mieloide crónica = 25-70 %.
7. Leucemia linfoide crónica = 15-35 %.
8. Linfoma de Hodgkin = 50 %.
Aunque la FC es una condición importante, no es suficiente para producir el SLT, ya que debe ir acompañada de una alta tasa de síntesis de purinas. Los tumores que agrupan ambas características son los primeros 6 de la lista anterior, por lo tanto, en ellos está indicada la prevención rutinaria del SLT. 13
Fisiopatología
Con la ruptura de las células tumorales se vierten al torrente sanguíneo aniones, cationes y productos del metabolismo, proteínas y ácidos nucleicos.
Hiperpotasemia: el potasio se encuentra en grandes cantidades en el medio intracelular y la ruptura masiva de las células provoca su salida al medio externo. La hiperpotasemia aparece en las primeras 6 a 72 horas, aunque inicialmente el riñón es capaz de eliminar el exceso de potasio. Otro factor que contribuye a la hiperpotasemia es la IRA, con lo cual el aumento del potasio se convierte en una complicación muy peligrosa que se manifiesta por debilidad muscular, parestesias y alteraciones del ritmo cardíaco.14
Hiperfosfatemia: las células tumorales contienen hasta 4 veces más fosfatos que las normales, por lo que su lisis provoca un aumento considerable de la concentración extracelular de fosfatos, que alcanza valores máximos a las 48-72 horas. El fosfato se une con el calcio y forma complejos de fosfato cálcico que precipitan en los túbulos y el intersticio renal y agravan la nefropatía pre-existente. A diferencia del ácido úrico, el fosfato cálcico disminuye su solubilidad a medida que aumenta el pH, sobre todo cuando es mayor de 8,0 y esto puede llegar a ser más peligroso que la hiperuricemia.14
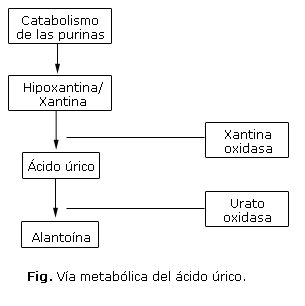
Hiperuricemia: es el evento más precoz y predispone al resto de las alteraciones, aparece entre las 48 a 72 horas de iniciada la quimioterapia. El catabolismo de las purinas desemboca en la formación de hipoxantina y xantina, que se transforma en ácido úrico por acción de la enzima xantina oxidasa (Fig.). El ácido úrico es un metabolito que disminuye su solubilidad a medida que se reduce el pH del medio y puede precipitar, lo que ocasiona una nefropatía obstructiva que desemboca en la insuficiencia renal aguda (IRA).14
Hipocalcemia: la concentración del calcio disminuye como resultado de la conjugación del calcio con el fósforo y constituye una de las manifestaciones más peligrosas del SLT. Los signos de Chvostek y Trousseau son expresión de la irritabilidad neuromuscular y preceden a los calambres musculares, la tetania y las arritmias cardíacas, que pueden ser letales.14
Insuficiencia renal aguda: se produce como consecuencia de la precipitación de los cristales de ácido úrico, fosfato cálcico y en menor medida xantinas que conducen a una nefropatía obstructiva aguda. Las manifestaciones relacionadas con esta complicación son la uremia, edemas, hipertensión, insuficiencia cardíaca y la exacerbación de los trastornos hidroelectrolíticos y metabólicos.14
Factores de riesgo
La probabilidad de presentar SLT y su severidad están en relación con factores dependientes del tumor y otros dependientes del paciente. Las neoplasias con gran masa tumoral y elevado recambio celular que son quimiosensibles y presentan valores elevados de lactato deshidrogenasa (LDH), tienen mayor riesgo de presentar un SLT grave. Además, si el paciente tiene alteraciones previas de la función renal, obstrucción urinaria, deshidratación o está utilizando medicamentos nefrotóxicos, está más expuesto a esta complicación.15
Diagnóstico
Los criterios para definir el SLT son heterogéneos. Recientemente se ha propuesto la clasificación de Cairo y Bishop que define 4 grados de SLT y distingue a los pacientes que solo tienen alteraciones humorales de los que presentan complicaciones clínicas graves (tabla 2). Estos criterios toman en consideración el grado de daño renal, la presencia de arritmias cardíacas y convulsiones.16,17
Criterios de laboratorio de SLT:
- Ácido úrico: > 476 mmol/L (8 mg/dL).
- Potasio: > 6,0 mmol/L (6 mEq/L).
- Fosfatos: > 2,1 mmol/L (niños) ó 1,45 mmol/L (adultos).
- Calcio: < 1,75 mmol/L.
El aumento mayor del 25 % con respecto al basal también se considera patológico. Se deben cumplir 2 o más criterios de laboratorio.
Criterios clínicos de SLT:
1. Insuficiencia renal: filtración glomerular menos de 60 mL/min, creatinina > 1,5 veces del valor normal para la edad.
2. Arritmias.
3. Convulsiones.
Los pacientes con SLT clínico deben tener al menos una manifestación además de los criterios de laboratorio del SLT.
Tanto para el diagnóstico como para el seguimiento de los pacientes con SLT, es necesario realizar química sanguínea completa, con especial énfasis en la evaluación de la función renal y el nivel de ácido úrico. El ionograma y la gasometría deben realizarse por lo menos 2 veces al día en las primeras 48-72 horas de iniciada la quimioterapia, pues permiten monitorizar los desequilibrios electrolíticos, sobre todo del potasio, el calcio y el fósforo. La medición del pH urinario es necesaria, sobre todo si se utiliza bicarbonato de sodio como parte del tratamiento. El paciente debe tener colocado un cardiomonitor para el diagnóstico precoz de los trastornos del ritmo cardíaco. El balance hídrico es recomendable para decidir el uso de diuréticos, sobre todo en los pacientes con alteraciones de la función cardíaca. 15,16
Tratamiento
La principal medida terapéutica del SLT es la prevención, que debe comenzar en las 48 horas previas al inicio de la quimioterapia. El oncohematólogo debe anticipar la ocurrencia a partir de las características del tumor, el tratamiento que se pretende utilizar y las condiciones físicas del paciente. En estos casos se iniciará una pre-fase citorreductora para evitar la lisis masiva de las células tumorales.
Hidratación: el aporte de líquidos debe ser abundante (3 000 mL/m2/día) para lograr un ritmo diurético de al menos 1,5 mL/kg/h. Debe comenzar 2 días antes del inicio de la quimioterapia y mantenerse hasta 2-3 días después de finalizado el tratamiento. Se debe garantizar un acceso venoso apropiado para el tratamiento enérgico de esta complicación. La hidratación amplia aumenta el volumen intravascular, ayuda a corregir los trastornos electrolíticos y reduce la concentración del potasio, fosfatos y ácido úrico. Además, aumenta el flujo sanguíneo renal, el filtrado glomerular y por lo tanto, el gasto urinario. La solución para hidratación debe ser hipotónica con dextrosa 5 %, cloruro de sodio 0,9 % y sobre todo, no debe contener potasio. Se debe tener cuidado con la sobrecarga de volumen en los pacientes de edad avanzada o con antecedentes de cardiopatías.14,15
Diuréticos: después de instaurar una hidratación adecuada, se debe valorar el uso de diuréticos. La furosemida (0,5-1 mg/kg/dosis cada 6 horas) es un diurético de asa que contribuye a la eliminación del potasio, pero es poco efectiva cuando los túbulos renales están afectados por la precipitación del ácido úrico; en estos casos, el manitol (0,5 g/kg en 15 min) es de mayor utilidad. La acetazolamida es un inhibidor de la anhidrasa carbónica que potencia la diuresis alcalina, por lo que se debe evitar en los pacientes con acidosis metabólica.13,15
Alcalinización de la orina: este resulta un punto controversial en el tratamiento del SLT. Para garantizar la solubilidad del ácido úrico debe alcanzarse un pH urinario entre 7,0-7,3. La alcalinización de la orina se puede lograr si se añade bicarbonato de sodio a la hidratación parenteral o si se utiliza acetazolamida. Sin embargo, esto puede aumentar la precipitación de las xantinas y el fosfato cálcico, sobre todo cuando el pH urinario es superior a 8,0. Además, el uso de bicarbonato aumenta la fuerza de unión del fosfato con el calcio lo que hace más severa la hipocalcemia. Debido a los efectos adversos del bicarbonato y la falta de evidencia del beneficio de la alcalinización de la orina, actualmente no se recomienda su empleo.15
Reducción de la hiperuricemia: tradicionalmente se ha utilizado el alopurinol en dosis de 300-500 mg/m2 o 10 mg/kg/día. El alopurinol es un análogo sintético de la hipoxantina, por lo que compite como sustrato de la enzima xantina oxidasa y bloquea la síntesis de ácido úrico. Este medicamento tiene varias desventajas pues, aunque disminuye la síntesis del ácido úrico, no elimina el que se encuentra preformado; por lo tanto, deben pasar al menos 48 horas antes de que se reduzca la hiperuricemia. Otra desventaja es la acumulación de metabolitos intermedios como hipoxantina y xantina, que tienen una solubilidad baja y pueden precipitar en el sistema excretor renal y producir una nefropatía por xantinas, que aumenta el riesgo de IRA. Además, interfiere con la degradación de la 6-mercaptopurina, 6-tioguanina y azatioprina, por lo que su dosis se debe reducir entre el 50 y 75 % si el paciente recibe alguno de estos agentes quimioterápicos. Su excreción renal obliga a ajustar la dosis en pacientes con IRA, sobre todo si aparecen manifestaciones adversas como rash, hepatitis, eosinofilia y empeoramiento renal.12-15
Desde hace 3 décadas se utiliza la enzima urato oxidasa que convierte el ácido úrico en alantoína, metabolito 5 a 10 veces más soluble. Posteriormente estuvo disponible en su forma recombinante bajo el nombre de Rasburicase. La dosis recomendada es de 0,15-0,2 mg/kg (en adultos de 6-8 mg/día) en infusión de 30 minutos, una vez al día. Este medicamento no se debe utilizar en los pacientes con déficit de glucosa 6 fosfato deshidrogenasa (G6PD), pues induce la hemólisis. La enzima urato oxidasa logra una disminución del 85 % del ácido úrico tras 4 horas de su administración, mientras el alopurinol solo logra el 12 %. Los pacientes tratados con esta enzima no requieren alcalinización de la orina, lo que facilita la excreción del fosfato cálcico. Actualmente se encuentra disponible la variante pegilada (PEG uricase) que aumenta la vida media y efectividad del producto.12,15
Tratamiento de la hiperpotasemia:
La alteración fundamental de la hiperpotasemia es la inducción de arritmias letales; por lo tanto, se requiere monitorización del paciente. Si la arritmia es inminente, se debe utilizar gluconato de calcio que estabiliza la membrana del cardiomiocito, aún a riesgo de aumentar la nefropatía por fosfatos. No obstante, esta es una solución temporal que debe acompañarse de medidas específicas para reducir el potasio extracelular.
Las resinas de intercambio iónico (Kayexalate) se pueden administrar por vía oral o por enemas, pero no logran una reducción inmediata del potasio. El bicarbonato de sodio solo es efectivo en los casos en que se acompañen de acidosis metabólica severa. Otras medidas a utilizar incluyen la combinación de dextrosa con insulina (dextrosa 10 %: 2 mL/kg + insulina simple 0,1 U/kg), los diuréticos de asa y la diálisis en los casos refractarios.13,15
Tratamiento de la hiperfosfatemia:
En los pacientes con aumento significativo de fosfatos que se acompañen de manifestaciones clínicas, se pueden utilizar quelantes de fósforo como el hidróxido de aluminio en dosis de 30 mL/ 6 horas. Las soluciones de dextrosa con insulina pueden ser efectivas. La diálisis está indicada en los pacientes con hiperfosfatemia severa o sintomática.14,16
Tratamiento de la hipocalcemia:
Los pacientes con disminución severa del calcio que se mantengan asintomáticos no deben ser tratados, sobre todo si tienen hiperfosfatemia, pues aumenta el riesgo de precipitación del fosfato cálcico y desarrollo de IRA. En los casos de irritabilidad neuromuscular manifiesta con signo de Chvostek o Trousseau positivos, tetania o arritmias, se administrará gluconato de calcio (50-100 mg/kg) con el objetivo de revertir las manifestaciones clínicas, no de corregir la hipocalcemia.14,15
Tratamiento de la IRA:
El reconocimiento precoz de la necesidad de diálisis antes que el deterioro del organismo sea irreversible, es la piedra angular del tratamiento de la IRA. Entre las modalidades de tratamiento dialítico (hemodiálisis, diálisis peritoneal, hemofiltración continua), no hay estudios comparativos que demuestren superioridad de alguna. No obstante, la hemodiálisis es superior a la diálisis peritoneal en la corrección de las alteraciones metabólicas. La eliminación del ácido úrico y los fosfatos es eficiente, pero es conveniente repetir el proceder cada 12 o 24 horas.
Los criterios de diálisis son:14,15
- Potasio: > 6 mEq/dL.
- Ácido úrico: > 900 mmol/L o 15 mg/dL.
- Creatinina: > 10 mg/dL.
- Fósforo: > 3,2 mmol/L o 10 mg/dL (o en rápido aumento).
- Hipocalcemia sintomática (tetania o convulsiones).
- Sobrecarga de volumen (fallo cardíaco) y oliguria.
A pesar del conocimiento de los factores de riesgo y las alteraciones patogénicas de esta urgencia oncológica, la mortalidad en los casos establecidos alcanza el 40 %. Por lo tanto, la prevención es la clave del tratamiento de estos pacientes, para minimizar las alteraciones metabólicas y el riesgo de daño renal agudo.16,17
En los humanos, esta vía metabólica solo llega hasta la formación de ácido úrico. La enzima urato oxidasa, presente de forma natural en otras especies animales, convierte el ácido úrico poco soluble en alantoína, hasta 10 veces más soluble.
HIPERLEUCOCITOSIS
Definición y etiología
Hiperleucocitosis se define como un aumento de leucocitos en sangre periférica mayor de 100 000/mm3, lo que es clínicamente significativo cuando sobrepasa los 200 000/ mm3 en leucemia mieloide aguda (LMA) y los 300 000/ mm3 en leucemia linfoide aguda (LLA).18 Cuando hay presencia masiva de blastos circulantes pueden sobrevenir urgencias debido a leucostasis en la vasculatura cerebral y pulmonar. En las leucemias linfoides agudas el incremento de blastos produce lisis tumoral; mientras que en las mieloides, ocasiona accidentes cerebrovasculares. Existen varios factores asociados con este cuadro según el tipo de leucemia (tabla 3).
La hiperleucocitosis se presenta entre el 9 y 14 % de las LLA, entre el 9 y 22 % de las LMA y en casi todas las leucemias mieloides crónicas en fase crónica. Las complicaciones causadas por este cuadro pueden ser la hemorragia o trombosis del sistema nervioso central (SNC), la leucostasis pulmonar y las alteraciones que acompañan al síndrome de lisis tumoral, las que se presentan con mayor morbilidad y mortalidad en las leucemia mieloides que en las linfoides (23 % vs. 5 %).18 Este cuadro de leucostasis generalmente está asociado con hiperleucocitosis, pero no siempre de forma invariable.
Patogenia
La hiperleucocitosis aumenta directamente la viscosidad sanguínea al aumentar el número de leucocitos, y también en forma indirecta por la mayor tendencia a formar agregados de células leucémicas y trombos leucocitarios. Como los blastos mieloides son de mayor tamaño que los linfoides, la hiperviscosidad es mayor en la LMA. Estas células se unen al endotelio gracias a la sobre expresión de moléculas de adhesión como la ICAM 1 y la P-selectina que inducen el daño pulmonar.19, 20
También se ha encontrado activación de otras moléculas de adhesión en el endotelio vascular de pacientes con LMA y leucostasis como CD54, CD62E, CD62P, CD106.21
Estos agregados leucocitarios dañan el endotelio de los vasos sanguíneos y producen hemorragias secundarias, las que pueden causar la muerte cuando ocurren en el SNC, corazón o pulmón. Los agregados leucocitarios al nivel pulmonar degeneran y liberan el contenido intracelular al espacio intersticial, lo que conduce a daño alveolar.22
Diagnóstico
Un paciente con una leucocitosis mayor que 100 000/ mm3 debe ser tratado rápidamente en un centro especializado para su correcta evaluación y tratamiento.
Se debe realizar: hemograma completo, electrolitos plasmáticos, pruebas de función renal y coagulograma, ya que el cuadro frecuentemente se asocia con coagulopatía, lo que aumenta aún más el riesgo de hemorragias.
La sintomatología está dominada por el compromiso neurológico y pulmonar, que se manifiesta con toma de conciencia variable, visión borrosa, diplopía, convulsiones, edema de papila, disnea, hipoxia y cianosis. Además, se describe priapismo y dactilitis.22 En la radiografía de tórax puede observarse un infiltrado pulmonar difuso.
Tratamiento
Como existe riesgo de lisis tumoral, el tratamiento debe ser instaurado rápidamente con hiperhidratación alcalina y alopurinol o urato oxidasa recombinante. Se debe evitar aumentar la viscosidad con transfusiones innecesarias (especialmente de glóbulos rojos) tratando de alcanzar niveles de hemoglobina normales. Si el recuento plaquetario es menor de 20 X 109/L, se deben transfundir plaquetas para disminuir el riesgo de hemorragias.
Se emplea la hidroxiurea en dosis de 50 a 100 mg/kg/ día, la que es capaz de reducir del 50 al 80 % el conteo de leucocitos en 24 a 48 horas, sin causar SLT ni empeorar la coagulopatía.23
La leucoféresis o la exanguinotransfusión (EXT) pueden disminuir rápidamente el número de leucocitos y mejorar la coagulopatía, aunque en algunos pacientes se ha visto un aumento reactivo del número de leucocitos después del proceder y tiene como inconvenientes la necesidad de un buen acceso venoso y la pérdida de plaquetas en el proceder. Generalmente se inician cuando hay más de 100 000/mm3 leucocitos en la LMA y más de 250 000/mm3 en la LLA, y se mantienen hasta que se reduzcan a 50 000/mm3 o menos.24,25
También se han usado drogas que interfieran con los receptores de adhesión y se ha comprobado que la dexametasona es capaz de inhibir la activación del CD 18, las L-selectinas y los receptores de la interleucina-8 en las células mieloides, y las E-selectinas e ICAM-1 en las células endoteliales.26,27
Además de todas estas medidas, se debe iniciar lo antes posible la quimioterapia citorreductora. En algunos centros se administra radioterapia de cráneo (400 cGy) para prevenir el riesgo de hemorragia de sistema nervioso central, aunque esta medida no es muy aceptada por todos y solo se usa como régimen de emergencia en caso de daño cerebral.28
SÍNDROME DE HIPERVISCOSIDAD
Definición
La hiperviscosidad es la resistencia de los líquidos a fluir libremente debido a la cohesión y adhesión de sus partículas. La viscosidad sanguínea (VS) es la resistencia de la sangre a fluir por los vasos sanguíneos y su incremento puede provocar un cuadro conocido como síndrome de hiperviscosidad (SHV).29
La hiperviscosidad se vincula con los elementos formes, en especial los eritrocitos, y las proteínas séricas. La correlación entre la VS y el hematócrito es lineal a cualquier concentración proteica. La VS también recibe la influencia de la agregación eritrocitaria, como ocurre durante la formación de pilas de moneda; el aumento de la viscosidad interna de los glóbulos rojos y la reducción de la flexibilidad o deformabilidad de los eritrocitos.30
La viscosidad del plasma depende del nivel y viscosidad intrínseca de las proteínas, que está supeditada a su vez al tamaño y configuración molecular, la interacción entre proteínas y células y a la naturaleza del lecho vascular. Como la IgM tiene un peso molecular alto y una configuración inusual, su viscosidad intrínseca es elevada, lo que explica la mayor frecuencia del SHV en la macroglobulinemia de Waldenström (MW). El componente monoclonal (M) IgA, también provoca hiperviscosidad por su tendencia a formar polímeros ligados por puentes disulfuro. La hiperviscosidad ligada al componente M IgG es menos común y generalmente se asocia con altas concentraciones.30, 31
Entidades relacionadas con el SHV29-31
Alteración sérica de proteínas y lipoproteínas.
- Hematológicas:
1. Macroglobulinemia de Waldenström.
2. Mieloma múltiple (MM).
3. Enfermedad de cadenas ligeras κ.
4. Crioglobulinemia.
- Reumatológicas:
1. Artritis reumatoide.
2. Síndrome de Sjögren.
3. Polimiositis-polisinovitis.
4. Lupus eritematoso sistémico.
- Endocrinas:
1. Hiperlipidemia.
2. Diabetes mellitus.
Alteraciones de las células sanguíneas.
- Aumento del número:
1. Policitemia primaria y secundaria.
2. Leucemias con hiperleucocitosis.
- Disminución de la adaptabilidad:
1. Diabetes mellitus
2. Drepanocitosis.
Fisiopatología
El SHV plasmática suele aparecer cuando la viscosidad del plasma supera los 4,0-5,0 unidades centipoise (cp) (normalidad: 1,6-2,5), aunque existe una gran variabilidad interpersonal entre los valores de viscosidad y el grado de afectación clínica. Así, por ejemplo, aunque la hiperviscosidad está presente en el 70 % de los pacientes con MW, la clínica solo se manifiesta en el 10-30 % de los casos. De manera similar, en el MM el SHV solo se manifiesta en el 4-8 % de los pacientes, a pesar de que la viscosidad generalmente oscila entre 2,0 y 3,0 cp.29
Manifestaciones clínicas
Los signos y síntomas del SHV se vinculan con alteraciones circulatorias provocadas por una mayor resistencia al flujo sanguíneo y se imbrican con las manifestaciones propias de la enfermedad de base que lo provocan. Este síndrome se caracteriza por una tríada de alteraciones oftalmológicas, neurológicas y diátesis hemorrágicas, aunque la afectación puede ser multisistémica.29-31 Entre las manifestaciones que se presentan están las siguientes:
- Manifestaciones generales: astenia, anorexia y fatiga.
- Oftalmológicas: alteraciones del fondo de ojo, tales como dilataciones y constricciones de las venas retinianas en "ristra de salchichas", exudados y hemorragias, visión borrosa y pérdida de la visión, diplopía, oclusión de las venas retinianas, y papiledema.
- Neurológicas: cefalea, mareos y vértigo. Nistagmo, ataxia, somnolencia, estupor y coma. Convulsiones, paresias, accidentes cerebrovasculares, dificultades en la concentración. Neuropatía periférica progresiva, mielopatía y miopatía, tinitus y sordera, por lo general secundaria a la trombosis de las venas del oído; agitación psicomotora, alucinaciones, inestabilidad emocional y desorientación.
- Cardiovasculares: disnea de esfuerzo, insuficiencia cardíaca congestiva, fenómeno de Raynaud, trombosis arterial, hipervolemia.
- Respiratorias: derrame pleural.
- Diátesis hemorrágicas: epistaxis, gingivorragias, equimosis, sangramiento digestivo, hematuria, alteraciones de la hemostasia.
- Nefrológicas y urológicas: proteinuria de Bence Jones secundaria a alteraciones de la función glomerular y tubular, insuficiencia renal (raro) y priapismo.
Diagnóstico de laboratorio
Viscosimetría: en algunos laboratorios se compara el lapso que requiere un volumen constante de líquido para fluir a través de un equipo llamado viscosímetro, con el tiempo que demora el agua. Se dispone además de una prueba rápida que usa una pipeta de glóbulos rojos como viscosímetro.
En los sujetos con viscosidad sérica relativa de 2 a 4 cp, casi nunca surgen problemas, pero sí en la mayoría con niveles de 5 a 8 y en casi todos cuando las cifras alcanzan de 8 a 10. Si los valores son superiores a 10, todos exhiben compromiso clínico.30
Tratamiento
- Plasmaféresis: todos los síntomas y signos del SHV se atenúan al menos de manera transitoria luego de este procedimiento.29-32 La paraproteína IgM es intravascular, por lo que una sola sesión de urgencia puede disminuir de manera considerable la sintomatología, pero por lo general y en el resto de las ganmapatías monoclonales, son necesarias varias sesiones y extraer la mitad del volumen plasmático o más.33,34 En cada sesión deben intercambiarse por lo menos de 2 a 4 litros de plasma cada 1 o 2 semanas.30
- Transfusión de sangre: la transfusión de sangre debe manejarse con precaución en estos pacientes, sobre todo en ancianos. El descenso de la masa eritrocitaria va acompañado del incremento del volumen plasmático de manera compensatoria, por lo tanto, la hiperviscosidad puede agravarse. Este factor, unido con la edad del paciente y la posibilidad de enfermedades subyacentes, aumenta el riesgo de insuficiencia cardíaca ante cualquier intento de corregir la anemia mediante la transfusión.30
- Tratamiento específico: solo el tratamiento de la enfermedad de base puede corregir de manera definitiva el estado de hiperviscosidad. Está indicado inicialmente el uso de esteroides como la prednisona en dosis de 20 mg cada 8 horas y quimioterapia en la MW y en el MM, unido con los avances actuales en el manejo de estas enfermedades.29,35,36
- Agentes quelantes: la penicilamina y otros agentes quelantes son capaces de disociar los agregados de inmunoglobulinas y reducen los puentes disulfuro.30
Múltiples son las urgencias que pueden surgir durante la evolución y tratamiento de las enfermedades hematológicas que obligan a mantener una actitud activa y alerta para diagnosticarlas y tratarlas rápidamente. De este modo, las secuelas pueden minimizarse, con lo que se evitan las complicaciones permanentes y se mejora la calidad de vida de los pacientes.
REFERENCIAS BIBLIOGRÁFICAS
1. Flombaum CD. Metabolic emergencies in the cancer patient. Semin Oncol. 2000 Jun;27(3):322- 34.
2. Solimando DA. Overview of hypercalcemia of malignancy. Am J Health Syst Pharm. 2001 Nov;15(8):34-7.
3. Morony S, Warmington K, Adamu S, Asuncion F, Geng Z, Grisanti M, et al. The inhibition of RANKL causes greater suppression of bone resorption and hypercalcemia compared with bisphosphonates in two models of humoral hypercalcemia of malignancy. Endocrinology. 2005 Aug;146(8):3235-43.
4. Liang M, Mallari C, Rosser M, Ng HP, May K, Monahan S, et al. Identification and characterization of a potent, selective and orally active antagonist of the CC chemokine receptor-1. J Biol Chem. 2000 Jun 23;275(25):190-8.
5. Beers MH, Berkow R, eds. The Merck Manual of Diagnosis and Therapy. 17th ed. Whitehouse Station, NJ: Merck Research Laboratories; 1999.
6. Terpos E, Sezer O, Croucher P. The use of bisphosphonates in multiple myeloma: recommendations of an expert panel on behalf of the European Myeloma Network. Ann Oncol. 2009 Aug;20(8):1303-17.
7. Dunford JE, Rogers MJ, Ebetino FH. Inhibition of protein prenylation by bisphosphonates causes sustained activation of Rac, Cdc42, and Rho GTPases. J Bone Miner Res. 2006 May;21(5):684-94.
8. Rosen LS, Gordon D, Antonio BS, Kaminski M, Howell A, Belch A, et al. Zoledronic acid versus pamidronate in the treatment of skeletal metastases in patients with breast cancer or osteolytic lesions of multiple myeloma: a phase III, double-blind, comparative trial. Cancer J. 2001 Sep-Oct;7(5):377-87.
9. Kraj M, Poglód R, Maj S, Pawlikowski J, Sokolowska U, Szczepanik J. Comparative evaluation of safety and efficacy of pamidronate and zoledronic acid in multiple myeloma patients (single center experience). Acta Pol Pharm. 2002 Nov-Dec;59(6):478-82.
10. Céspedes-Quevedo MC, Ropero-Toirac R, Torres-Linares M. Estimación del filtrado glomerular por cálculo, utilizando la creatinina sérica. Bol Cientif ISCM (Santiago de Cuba). 1994;6(1):159-68.
11. Madan J, Arrowsmith R. Complications of Hematopoietic Neoplasms. En: Greer JP, Lukens JN eds. Wintrobe's Clinical Hematology. 11th ed. Philadelphia: Lippincott Williams & Wilkins Publishers; 2003.
12. Goldman SC, Holcenberg JS, Finklestein JZ, Hutchinson R, Kreissman S. A randomized comparison between rasburicase and allopurinol in children with lymphoma or leukemia at high risk for tumor lysis Blood. 2001 May 15;97(10):2998-3003.
13. Rizzardini C, Espinoza X. Urgencias oncológicas. Rev Ped Elec (en línea) 2005;2(2):25-32. ISSN 0718-0918. Disponible en: http://www.revistapediatria.cl/vol2num2/pdf /8_urgencias_oncologicas.pdf
14. Coiffier B, Altman A, Pui CH, Younes A, Cairo MS. Guidelines for the management of pediatric and adult tumor lysis syndrome: an evidence-based review. J Clin Oncol. 2008 Jun 1;26(16):2767-78.
15. Hochberg J, Cairo M. Tumor lysis syndrome: current perspective. Haematologica. 2008 Jan;93(1):67-74.
16. Rampello E, Fricia T, Malaguarnera M. The management of tumor lysis syndrome. Nat Clin Pract Oncol. 2006 Aug;3(8):438-47.
17. Tosi P, Barosi G, Lazzaro C, Liso V, Marchetti M, Morra E, et al. Consensus conference on the management of tumor lysis syndrome. Haematologica. 2008 Dec;93(12):1877-85.
18. Porcu P, Cripe LD, Ng EW, Bhatia S, Danielson CM, Orazi A, et al. Hyperleukocytic leukemias and leukostasis: A review of pathophysiology, clinical presentation and management. Leuk Lymphoma. 2000 Sep;39(1-2):1-18.
19. Litchmann MA, Heal J, Rowe JM. Hyperleukocytic leukemia: rheological and clinical features and management. Baillieres Clin Haematol. 1987 Sep;1(3):725-46.
20. Doerschuck CM, Quinlan WM, Doyle NA, Bullard DC, Vestweber D, Jones ML, et al. The roles of P-selectin and ICAM-I in acute lung injury as determined using anti-adhesion molecule antibodies and mutant mice. J Immunol. 1996 Nov 15;157(10):4609-14.
21. Kamasaka T, Quinlan WM, Doyle NA, Condon TE , Sligh J, Takei F, et al. The role of ICAM-1 in endotoxin-induced pneumonia evaluated using ICAM- 1 antisense, anti-ICAM-1 antibodies and ICAM-1 mutant mice. J Clin Invest. 1996 May 15;97(10):2362-9.
22. Stucki A, Rivier AS, Gikic M, Monai N, Schapira M, Spertini O. Endothelial cell activation by myeloblasts: molecular mechanisms of leukostasis and leukemic cell dissemination. Blood. 2001 Apr 1;97(7):2121-9.
23. McKee LC, Collins R D. Intravascular thrombi and aggregates as a cause of morbidity and mortality in leukemia. Medicine (Baltimore). 1974 Nov;53(6):463-78.
24. Grund EM, Armitage JO, Bums CP. Hydroxyurea in the prevention of the effects of leukostasis in acute leukemia. Arch Intern Med. 1977 Sep;137(9):1246-7.
25. Bug G, Anargyrou K, Tonn T, Bialleck H, Seifried E, Hoelzer D, et al. Impact of leukapheresis on early death rate in adult acute myeloid leukemia presenting with hyperleukocytosis. Transfusion. 2007 Oct;47(10):1843-50.
26. Karp D, Beck JR, Comell CJ Jr. Chronic granulocytic leukemia with respiratory distress. Efficacy of emergency leukapheresis. Arch Intern Med. 1981 Sep;141(10):1353-4.
27. Filep JG, Delalandre A, Payette Y, Foldes-Filep E. Glucocorticoid receptor regulates the expression of L-selectin and CDllICD18 on human neutrophils. Circulation. 1997 Jul 1;96(1):295-301.
28. Chang MC, Chen TY, Tang JL, Lan YJ, Chao TY, Chiu CF, et al. Leukapheresis and cranial irradiation in patients with Hyperleukocytic acute myeloid leukemia: No impact on early mortality and intracranial hemorrhage. Am J Hematol. 2007 Nov;82(11):976-80.
29. Gómez E, Roncero C, De Pablo J, Rovira M. Síndrome de hiperviscosidad y alteraciones mentales. Acta Esp Psiquiatr. 2000;28(4):263-6.
30. Foerster J. Discrasias de células plasmáticas: consideraciones generales. Hematología Clínica de Wintrobe. 9na. ed. Buenos Aires: Editorial Intermédica; 1995. pp. 1909-23.
31. Seth R, Bhat AS. Management of Common Oncologic Emergencies. Indian J Pediatr. 2011 Mar 12. [Epub ahead of print] DOI 10.1007/s12098-011-0381-5.
32. Mullen E, Méndez N. Hyperviscosity syndrome in patients with multiple myeloma. Oncol Nurs Forum. 2008 May;35(3):350-2.
33. Zojan N, Ludwig H. Hematological emergencies. Ann Oncol. 2007 Jan;18(1):45-8.
34. Ballestri M, Ferrari F, Magistroni R, Mariano M, Ceccherelli GB, Milanti G, et al. Plasma exchange in acute and chronic hyperviscosity syndrome: a rheological approach and guideline study. Ann Ist Super Sanita. 2007;43(2):171-5.
35. Palumbo A, Rajkumar SV. Treatment of newly diagnosed myeloma. Leukemia. 2009 Mar;23(3):449-56.
36. Kyle RA, Rajkumar SV. Treatment of multiple myeloma: a comprehensive review. Clin Lymphoma Myeloma. 2009 Aug;9(4):278-88.
Recibido: 13 de septiembre del 2011.
Aprobado: 11 de octubre del 2011.
Dr. Juan Carlos Jaime-Fagundo. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800. La Habana, Cuba. Tel (537) 643 8695, 8268, Fax (537) 644 2334. Correo electrónico: ihidir@hemato.sld.cu Website: http://www.sld.cu/sitios/ihi


{kind=link}
{kind=link}