










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión impresa ISSN 0864-0289
Rev Cubana Hematol Inmunol Hemoter vol.28 no.4 Ciudad de la Habana oct.-dic. 2012
ARTÍCULO DE REVISIÓN
Regulación de la hepcidina y homeostasis del hierro: avances y perspectivas
Regulation of hepcidin and iron homeostasis: progress and prospects
MSc. Mariela Forrellat-Barrios, Dra. Norma Fernández-Delgado, Prof. Dr.Cs. Porfirio Hernández-Ramírez
Instituto de Hematología e Inmunología. La Habana, Cuba.
RESUMEN
El estudio de los desórdenes genéticos del metabolismo del hierro, la identificación de sus transportadores y el descubrimiento de la hepcidina, hormona reguladora de la homeostasia del hierro, han contribuido grandemente a aumentar los conocimientos sobre este metabolismo y han cambiado sustancialmente la visión sobre las enfermedades relacionadas con alteraciones del metabolismo férrico. En la última década, no solo se han esclarecido elementos de la patogénesis de estas enfermedades, sino que ya se vislumbran aplicaciones terapéuticas de estos avances. Así, ya se habla de una nueva era basada en el tratamiento de los desórdenes de la homeostasia del hierro a través de la modulación de la hepcidina.
Palabras clave: hierro, hepcidina, ferroportina, homeostasia del hierro.
ABSTRACT
The study of genetic disorders of iron metabolism, identification of transporters and the discovery of hepcidin- a hormone regulating iron homeostasis- have contributed greatly to increase awareness of this metabolism. Substantially, the vision on diseases related to disorders of iron metabolism has been changed. In the last decade, elements of the pathogenesis of these diseases have not only been clarified, but therapeutic applications of these advances are looming. Thus, there are expectations of a new era based on the treatment of iron homeostasis disorders through hepcidin modulation.
Keywords: iron, hepcidin, ferroportin, iron homeostasis.
INTRODUCCIÓN
Los últimos años se han caracterizado por una gran actividad investigativa en el campo del metabolismo del hierro. Como resultado, se han producido importantes avances en el esclarecimiento de los mecanismos de control de la homeostasia del mineral, especialmente en lo referido a la regulación de la expresión de la hepcidina. Igualmente se ha avanzado notablemente en el conocimiento de la fisiopatología de enfermedades relacionadas con las alteraciones del metabolismo férrico, lo que ha permitido plantear nuevas posibilidades terapéuticas para su manejo.
Para asegurar un suministro de hierro adecuado para las funciones celulares esenciales sin provocar toxicidad por hierro, requiere que sus concentraciones en plasma y fluidos extracelulares se mantengan entre 10-30 µmol/L. Desde hace algo más de una década se conoce que la hepcidina es un elemento central en la regulación del flujo de hierro al plasma, ya sea a partir de la absorción del mineral presente en la dieta (1-2 mg), el reciclaje de los eritrocitos (20 mg/d) o la movilización de las reservas en hígado y macrófagos (algunos mg/d de acuerdo con las necesidades).1,2
REGULACIÓN DE LA ABSORCIÓN DE HIERRO
Desde la primera mitad del siglo XX se planteó que el control de la homeostasia del hierro se produce por regulación de su absorción. Se conoce que la tasa de absorción es afectada por una serie de factores, algunos interrelacionados entre sí, que pueden actuar simultáneamente, como son: el estado de las reservas corporales, la hipoxia, la actividad eritropoyética y la inflamación. El proceso de absorción ocurre casi exclusivamente en el duodeno y sigue una secuencia de pasos. En él están implicadas una serie de proteínas que forman parte de la compleja red molecular del metabolismo del hierro.2,3
El primer paso es la reducción del hierro del estado férrico a ferroso. Esto ocurre por la actividad ferri-reductasa del citocromo b duodenal al nivel del borde en cepillo del enterocito. Una vez en estado ferroso, el hierro es transportado a través de la membrana plasmática al interior de la célula por el transportador de metales divalentes (DMT 1). Dentro del enterocito, el mineral puede tener 2 destinos: ser almacenado como ferritina y excretado en las heces cuando se produce la decamación de los enterocitos senescentes, o puede ser transferido por la ferroportina (Fpn) a través de la membrana basolateral al plasma, proceso en el que se requiere la actividad ferroxidasa de la hefastina. La expresión de cada una de las proteínas involucradas está sujeta a regulación; así, de acuerdo con las necesidades de hierro del organismo, la expresión de DMT1 y Fpn son moduladas para regular la cantidad de Fe que se absorbe y pasa a la circulación.2-5
Para explicar la regulación de la absorción se han propuesto 2 modelos: el de las criptas programadas y el de la hepcidina.
Modelo de las criptas programadas
Plantea que la cantidad de hierro que se absorbe viene determinada por la carga de hierro de los enterocitos en las criptas. La expresión de DMT1, del receptor de transferrina 1 (TfR1) y de la ferritina, son controladas por los niveles de hierro en estas células a través de la interacción de las proteínas reguladoras de hierro (IRP1 y 2) con los elementos de respuesta al hierro (IRE) en los ARN mensajeros (ARNm) de estas proteínas. En ausencia de hierro, la IRP1 se une con el IRE del ARNm del TfR1, DMT1 y Fpn, el transcripto se estabiliza, se produce la traducción y en consecuencia, las proteínas son sintetizadas. Luego, el aumento de la actividad de unión de IRP1 es reflejo de la disminución de las reservas corporales de hierro que resulta en la estimulación de la síntesis de estas proteínas con el consecuente aumento de la absorción intestinal de hierro. Sin embargo, cuando las IRP se unen al IRE del ARNm de la ferritina la traducción del transcripto se bloquea y la síntesis se detiene. Así, las concentraciones de ferritina son recíprocamente reguladas, estando aumentadas en los estados de repleción y disminuidas en la depleción del mineral.2,4-6
Modelo de la hepcidina
Plantea que el centro de control del tráfico de hierro es el hígado y su efector la hepcidina, pequeña hormona peptídica que se produce fundamentalmente por los hepatocitos en respuesta a la cantidad de hierro circulante, cuya expresión es modulada por los mismos factores que la absorción del mineral. Esta proteína es secretada a la circulación e interactúa con los enterocitos vellosos donde regula el nivel de absorción de hierro a través del control de la expresión de la Fpn en la membrana basolateral de estas células.3,6
La unión de la hepcidina a la Fpn provoca inicialmente la fosforilación de tirosina mediada por la Janus kinasa 2 (JAK 2) del lazo citosólico de la proteína transportadora. La proteína fosforilada es entonces internalizada, desfosforilada, ubiquitinada y finalmente degradada en el compartimiento endosómico/ lisosomal. Más tarde, si se requiere hierro en la médula ósea para la síntesis de hemoglobina, la Fpn es rexpresada en la superficie celular y la liberación de hierro a la circulación se reanuda. Las moléculas de Fpn presentes en los macrófagos y hepatocitos son también diana para la hepcidina.7,8
Este modelo demuestra que la homeostasia del hierro depende de la retroalimentación regulatoria entre necesidades y reservas. Además, coincide con lo planteado en la década de 1950 acerca de la existencia de una señal entre el compartimiento de reservas (macrófagos y hepatocitos) y los sitios de utilización (fundamentalmente médula ósea) que determina la cantidad de hierro liberada a la circulación y entregada a la eritropoyesis.3,8
Las concentraciones de hierro plasmático y la saturación de transferrina van a reflejar la diferencia entre el hierro transferido al plasma, regulado por la interacción hepcidina/Fpn, y el consumo del mineral en la médula ósea por la actividad eritropoyética; y en menor grado, por otros tejidos. El compartimiento plasmático de transferrina es relativamente pequeño y puede ser recambiado cada 3 horas, aproximadamente. De este modo, las concentraciones de hierro responden rápidamente a los cambios en las concentraciones de hepcidina.2,3
La incorporación celular de hierro en sus diferentes formas (hierro elemental o hierro hemo por los enterocitos, transferrina diférrica, complejo hemo-hemopexina, hemoglobina-haptoglobina, eritrocitos senescentes por macrófagos) son también regulados, pero parece ser que la regulación de la expresión de Fpn en la membrana celular, es el modo predominante a través del cual se regula la transferencia de hierro al plasma.1
REGULACIÓN DE LA EXPRESIÓN DE LA HEPCIDINA
Como se ha planteado, mantener un suministro constante de hierro a la médula ósea para la eritropoyesis es de máxima prioridad. Así, cuando las demandas se incrementan en el compartimiento eritropoyético, la salida de hepcidina a la circulación debe ser disminuida. La expresión de la hepcidina es modulada por los mismos factores que modifican la absorción de hierro y controlada por múltiples vías de señalización que incluyen señales inhibitorias y estimuladoras. Entre las que inhiben están la disminución del hierro sérico y hepático, la anemia, la hipoxia, los factores derivados de la médula ósea (GDF15, growth diferentiation factor15 y TWSP1, twisted gastrulation protein 1) y la eritropoyetina. Las estimuladoras incluyen el aumento del hierro sérico y hepático, las citocinas inflamatorias, las BMP (del inglés bone morphogenetic protein) y el estrés en el retículo endoplásmico.2,8
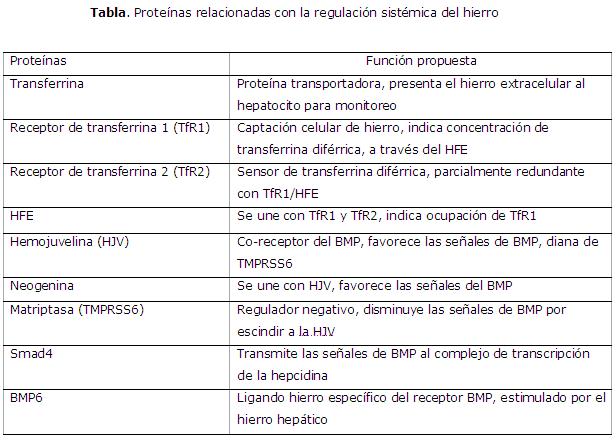
La regulación de la producción de hepcidina ocurre al nivel transcripcional, aunque hay evidencias de otro tipo de control. En ella participa una serie de moléculas implicadas en el metabolismo del hierro (tabla). Todas estas proteínas son susceptibles de sufrir mutaciones que pueden provocar la deficiencia de hepcidina y la consecuente sobrecarga de hierro, o la sobrexpresión de la hormona con la consiguiente deficiencia y secuestro del mineral en los macrófagos.3,9
Regulación mediada por hierro
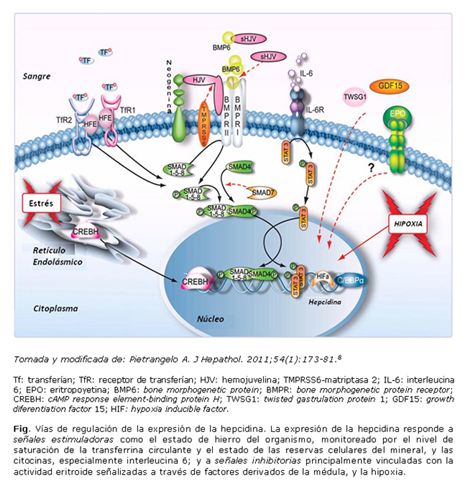
Comprender la regulación de la hepcidina por el hierro es cada día más difícil por la aparente redundancia del sistema3 (Fig.). El mecanismo regulatorio de respuesta al hierro se centra en las BM, su receptor BMPR y su vía de señalización intracelular a través de las Smad (del inglés, small mothers against decapentaplegic).10
La presencia de hierro induce la producción de BMP6, ligando cuya única función conocida es la regulación de la expresión de la hepcidina,11,12 y el subsecuente ensamblaje de un complejo heterotetramérico anclado a membrana con actividad serin/treonina kinasa que actúa como receptor (BMPR). Se produce la activación de la cascada de señalización común que involucra la fosforilación del receptor intracelular activado Smad 1,5 y 8, que interactúan en el citoplasma con la Smad 4. El complejo resultante se transloca al núcleo donde activa la transcripción del gen de la hepcidina. Por su parte, la Smad 7, también estimulada por hierro, parece atenuar la señal de activación de la hepcidina.1,8
La hemojuvelina (HJV) anclada a membrana actúa como un co-receptor del BMPR y su expresión en membrana es modulada por otras 2 proteínas: la matriptasa 2 (conocida por las siglas de su gen TMPRSS6), que degrada la HJV en respuesta al hierro y así actúa como regulador negativo de la vía del BMP.13 La neogenina, proteína que interactúa con la HJV y el BMPR, aunque su vínculo real con la regulación del hierro aún no está bien establecido.14
Otros sensores que participan en esta vía de señalización son las concentraciones de transferrina diférrica, las concentraciones de TfR 1 y 2 y su contraparte transmembrana la proteína HFE. El incremento de las concentraciones de transferrina diférrica cambia la interacción de HFE del TfR1 al TfR2, con lo que promueve la estabilización de esta proteína y estimula la señal Smad.1,2,14
Regulación mediada por la inflamación
Durante la infección y la inflamación se generan señales que incrementan drásticamente la síntesis y liberación de hepcidina, con la consiguiente hipoferremia, eritropoyesis restringida en hierro y anemia.1 Una de las citocinas responsables de este efecto es la IL-6, interleucina que activa la vía JAK-STAT (Fig.). Después de la fosforilación por la JAK2, el factor de transcripción STAT3 se une a regiones conocidas del promotor de la hepcidina, con lo que incrementan su transcripción.1,15 Otras citocinas inflamatorias y productos microbianos ejercen igualmente su función en la activación de la transcripción de la hepcidina. Se ha planteado también una posible interacción con la vía BMP/Smad.8,16,17
El estrés del retículo endoplásmico, estado celular derivado de la acumulación de proteínas inadecuadamente plegadas, también induce expresión de hepcidina por un programa conocido como UPR (unfolded protein response). El UPR es también activado por lipopolisacáridos y citocinas como IL-6 y IL-1ß, lo que sugiere que la inducción de hepcidina por el estrés del retículo endoplásmico debe cooperar con la respuesta vía JAK/STAT. Otro elemento que se ha planteado que contribuye a la respuesta de la hepcidina al estímulo inflamatorio es la CREBH (cAMP response element-binding protein H), factor transcripcional que es activado durante el estrés del retículo endoplásmico para promover la producción de proteínas de fase aguda.17,18
Regulación por factores eritroides
En la anemia por deficiencia de hierro, en las anemias hereditarias con eritropoyesis ineficaz y en los modelos animales de anemia por sangramiento o hemólisis, se han observado concentraciones bajas de hepcidina. Todo ello indica que existe una señal reguladora de la hepcidina que se origina en los precursores eritroides de la médula ósea. Entre los factores propuestos están el GDF 15 y el TWSG1, ambos miembros de la superfamilia de los factores transformadores de crecimiento ß (TGF-ß).2,19
La expresión de la hepcidina es también modulada por la hipoxia, la que induce la producción de eritropoyetina por el riñón y por lo tanto, el incremento de la actividad eritropoyética.17 Sin embargo, la hipoxia también puede ejercer un efecto local sobre la producción hepática de la hepcidina a través de la acción del HIF (del inglés, hypoxia inducible factor), factor de transcripción que promueve la expresión de genes que median la respuesta a la hipoxia. No obstante, se ha planteado que es posible que la hipoxia pueda afectar la producción de hepcidina localmente en el hígado por otros mecanismos como son el aumento de los niveles de HJV soluble, el incremento de la expresión de la TMPRSS6 o ambos.20,21
Aclaramiento de la hepcidina
Por ser un péptido pequeño no unido fuertemente a proteínas plasmáticas, la hepcidina es eliminada del plasma por el riñón y por endocitosis mediada por Fpn y proteólisis. De acuerdo con esto, las concentraciones de hepcidina pueden también incrementarse por enfermedades que disminuyan el aclaramiento renal de la proteína.1,22
HEPCIDINA Y DESÓRDENES DEL METABOLISMO DEL HIERRO
Las alteraciones de la regulación de la hepcidina son responsables de los desórdenes del metabolismo del hierro. En general, se producen 2 fenotipos totalmente opuestos: sobrecarga y restricción de hierro.
Desórdenes con sobrecarga de hierro
En ellos hay una hiperactividad de la Fpn que estimula la absorción intestinal de hierro y su liberación por los macrófagos, lo que causa el aumento de las concentraciones plasmáticas de hierro, de la saturación de transferrina y el depósito del exceso de hierro en el hígado y otros órganos, con el consiguiente daño.1,2
Este es el caso de la hemocromatosis hereditaria tipo I donde, debido a la pérdida de la actividad funcional de la HFE, se produce la interrupción de la vía de señalización BMP/Smad con lo que se atenúa la transcripción de la hepcidina y se produce la consecuente sobrecarga plasmática de hierro y el daño por toxicidad en los órganos donde se acumula.8
Otro ejemplo son las anemias con eritropoyesis ineficaz, como la ß talasemia y las anemias diseritropoyéticas congénitas, en las que la producción de hepcidina es suprimida por una señal patológica de los precursores eritroides incapaces de madurar a eritrocitos totalmente maduros. La señal tal vez sean los factores eritroides inducidos por la eritropoyesis ineficaz GDF15 y TWSG1, que como se ha planteado, inhiben las transcripción de la hepcidina con lo que causan liberación irrestricta del hierro a la circulación.2,8
Desórdenes restringidos en hierro
Se caracterizan por excesivas concentraciones de hepcidina plasmática debido a la estimulación de su producción por la inflamación sistémica, la producción autónoma por tumores o por mutación de la TMPRSS6 o la disminución de su aclaramiento por enfermedad renal. Independientemente de la causa específica, las concentraciones aumentadas de hepcidina inhiben la absorción de hierro en el duodeno y restringen su liberación por los macrófagos, lo que limita la disponibilidad de hierro para la síntesis de hemoglobina y resulta en las anemias restringidas en hierro.1 La hiperhepcidinemia prolongada conduce al secuestro del hierro en los macrófagos y anemia restringida en hierro. A pesar de la disminución del hierro sérico, se produce un aumento de la ferritina sérica como consecuencia del exceso de hierro tisular y de los mediadores inflamatorios.8
Otros mecanismos, como el acortamiento de la vida media de los eritrocitos, la supresión directa de la eritropoyesis por la citocinas y la disminución de la producción de eritropoyetina, pueden también contribuir a la anemia en esos casos, en dependencia de la enfermedad de base.1
TERAPIA DE HEPCIDINA
Basado en lo que se conoce sobre la biología y regulación de la hepcidina, se puede predecir que cualquier factor genético o adquirido que disminuya la síntesis o actividad de esta proteína conducirá a un flujo irrestricto de hierro a la circulación, a lo que seguirá la sobrecarga tisular con su potencial toxicidad y posibilidad de daño.8 En estas condiciones sería beneficioso el uso de terapias con hepcidina o sus agonistas, agentes que remplazan la actividad de la hormona o estimulan su producción endógena con lo que pueden prevenir la acumulación de hierro.1
Aunque estas estrategias no revertirían la sobrecarga de hierro, podrían disminuir el daño tisular mediado por hierro al redistribuir el hierro del parénquima a los macrófagos donde es menos tóxico.1
Por otra parte, los factores genéticos o adquiridos que causan aumento de la síntesis de hepcidina provocan la disminución de la transferencia de hierro al plasma y por tanto, la hipoferremia. Si la estimulación persiste, la eritropoyesis restringida en hierro y la anemia se instalan.8 En estos casos tendrían aplicación los antagonistas de la hepcidina, moléculas que disminuyen la producción de hepcidina o interfieren con su efecto sobre la Fpn. Esto aliviaría la restricción de hierro y provocaría la liberación del mineral para la eritropoyesis. En principio, los antagonistas pueden neutralizar las citocinas estimuladoras de hepcidina (BMP, IL-6), estimular la eritropoyesis, unirse y neutralizar el péptido hepcidina (anticuerpos y otras moléculas de unión), prevenir la unión de la hepcidina a la Fpn o interferir con la vía de internalización de esta última.1
Una nueva era se avizora en el tratamiento de los desórdenes del metabolismo del hierro, en la que se pasaría de los tratamientos con flebotomías para remover el exceso de hierro y los suplementos para repletar las reservas, a movernos dentro de las células y organelos. Ello sin duda abrirá nuevos horizontes para tratar o complementar el manejo de las enfermedades asociadas a alteraciones del metabolismo del hierro.8
Si bien en la actualidad ya han sido identificados los mecanismos de regulación del balance de hierro sistémico por el eje hepcidina-Fpn, los que regulan el balance de hierro celular, y se ha planteado que ambas vías interactúan para lograr la homeostasia de hierro en el organismo, aún quedan interrogantes y nuevos elementos por descubrir. Investigaciones recientes demostraron que bajo ciertas circunstancias, la regulación hepática de la expresión de hepcidina no puede compensar totalmente los defectos en el balance celular del hierro en los enterocitos para mantener el balance de hierro sistémico.23 Muy recientemente se ha planteado que la expresión de hepcidina puede ser modulada por microARN (miARN), lo que adiciona nuevas complejidades a la regulación de la homeostasia del hierro.24 Los miARN son una clase de ARN cortos que no codifican y que se unen con secuencias complementarias en los transcriptos diana para regular negativamente la expresión de los genes a nivel postranscripcional.17 Ello demuestra que, a pesar de lo mucho que se ha avanzado en la última década, aún queda mucho por andar en el camino de la ferrobiología.
REFERENCIAS BIBLIOGRÁFICAS
1. Ganz T, Nemeth E. The hepcidin-ferroportin system as a therapeutic target in anemias and iron overload disorders. Hematology Am Soc Hematol Educ Program. 2011;2011:538-42.
2. Fleming RE, Pomka P. Iron overload in human disease. New Engl J Med. 2012 Jan;366(4): 348-59.
3. Ganz T. Hepcidin and iron regulation, 10 years later. Blood. 2011 Apr;117(17):4425-33.
4. Forrellat-Barrios M, Fernández-Delgado N, Hernández-Ramírez P. Nuevos conocimientos sobre el metabolismo del hierro. Rev Cubana Hematol Inmunol Hemoter. 2005 Dic; 21(3): Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892005000300003&lng=es
5. Zhang AS, Enns CA. Molecular mechanisms of normal iron homeostasis. Hematology Am Soc Educ Program. 2009:207-14.
6. Muñoz M, García-Erce JA, Remacha AF. Disorders of iron metabolism. Part 1: Molecular basis of iron homeostasis. J Clin Pathol. 2011 Apr;64(4):281-6.
7. Ramey G, Deschemin JC, Durel B, Canonne-Hergaux F, Nicolas G, Vaulont S. Hepcidin targets ferroportin for degradation in hepatocytes. Haematologica. 2010 Mar;95(3):501-4.
8. Pietrangelo A. Hepcidin in human iron disorders: therapeutics implications. J Hepathol. 2011 Jan;54(1):173-81.
9. Flanagan JM, Truksa J, Peng H, Lee P, Beutler E. In vivo imaging of hepcidina promoter stimulaton by iron and inflammation. Blood Cell Mol Dis. 2007 May-Jun;38(3):253-7.
10. Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, Samad TA, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidina expression. Nat Genet. 2006 May; 38(5):531-9.
11. Meynard D, Kautz L, Darnaud V, Canonne-Hergaux F, Coppin H, Roth MP. Lack of the bone morphogenetic protein BMP6 induces iron overload. Nat Genet. 2009 Apr;41(4):478-81.
12. Andriopoulos B Jr, Corradini E, Xia Y, Faasse SA, Chen S, Grgurevic L, et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet. 2009 Apr; 41(4):482-7.
13. Silvestri L, Pagani A, Nai A, De Domenico I, Kaplan J, Camaschella C. The serine protease matriptase 2 (TMPRSS6) inhibits hepcidina activation by cleaving membrane hemojuvelin. Cell Metab. 2006 Dec;8(6):502-11.
14. Goswani T, Andrews NC. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for Mammalian iron sensing. J Biol Chem. 2006 Sep;281(39):28494-8.
15. Wrighting DM, Andrews NC. Interleukin-6 induces hepcidina expression through STAT3. Blood 2006 Nov;108(9):3204-9.
16. Babitt JL, Huang FW, Xia Y, Sidis Y, Andrews NC, Lin HY. Modulation of bone morphogenetic proteins signaling in vivo regulates systremic iron balance. J CLin Invest. 2007 Jul;117(7):1933-9.
17. Finberg KE. Unraveling mechanisms regulating systemic iron homeostasis. Hematology. Am Soc Hematol Educ Program. 2011;2011:532-7.
18. Vecchi C, Montosi G, Zhang K, Lamberti I, Duncan SA, Kaufman RJ, et al. ER stress controls iron metabolism through induction of hepcidina. Science. 2009 Aug;325(5942):877-80.
19. Ganz T, Nemeth E. Hepcidin and disorders of iron metabolism. Annu Rev Med. 2011;62:347-60.
20. Silvestri L, Pagani A, Camascella C. Furin-mediated release of soluble hemojuvelin: a new link between hypoxia and iron homeostasis. Blood. 2008 Jan;111(2):924-31.
21. Lakhal S, Schodel J, Towsend AR, Pugh CW, Ratcliff PJ, Mole DR. Regulation of type II transmembrane serin proteinase TMPRSS6 by hypoxia-inducible factors: a new link between hypoxia signalling and iron homeostasis. J Biol Chem. 2011 Feb;286(6):4090-7.
22. Zanitsky J, Young B, Wang HJ, Westerman M, Olbina G, Nemeth E, et al. Hepcidin a potential novel biomarker for iron status in chronic kidney disease. Clin J Am Soc Nephrol. 2009 Jun;4(6):1051-6.
23. Hentze MW, Muckenthaler MV, Galy B, Camaschella C. Two to tango:regulation of mammalian iron metabolism. Cell. 2010 Jul;142(1):24-38.
24. Castoldi M, Vujic Spasic M, Altamura S, Elmén J, Lindow M, Kiss J, et al. The liver-especific microRNA miR-122 controls systemic iron homeostasis in mice. J Clin Invest. 2011;121(4):1386-96.
Recibido:15 de agosto del 2012.
Aprobado: 15 de agosto del 2012.
MSc. Mariela Forrellat Barrios. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800, La Habana, Cuba. Tel (537) 643 8695, 8268, Fax (537) 644 2334. Correo electrónico: rchematologia@infomed.sld.cu
Website: http://www.sld.cu/sitios/ihi

{kind=link}
{kind=link}