










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión impresa ISSN 0864-0289
Rev Cubana Hematol Inmunol Hemoter vol.28 no.4 Ciudad de la Habana oct.-dic. 2012
COMUNICACIÓN BREVE
Cuantificación de hemoglobina A2 por electroforesis en gel de agarosa
Quantitation of hemoglobin A2 by electrophoresis in agar gel
Lic. Maydelín Miguel-Morales,I Lic. Liz Mabel Díaz-Barroso,I Dra. Heidys Garrote-Santana,I Téc. Grisel Uley-del Rosario,I Téc. Graciela Pérez-Diez de los Ríos,I Dra.Cs. Marianela Estrada-del CuetoI
I Instituto de Hematología e Inmunología. La Habana, Cuba.
INTRODUCCIÓN
En las distintas etapas del desarrollo -desde el embrión hasta el individuo adulto- diferentes hemoglobinas se sintetizan en un orden fijado y rigurosamente determinado. Al momento del nacimiento, aunque en diferentes concentraciones, son 3 las hemoglobinas presentes: hemoglobina (Hb) A, Hb fetal (Hb F) y Hb A2.1
La Hb A2 es una proteína compuesta por 2 cadenas polipeptídicas alfa (a) y 2 delta (d ) que dan lugar a la formación del tetrámero a2 d2. La activación del gen que codifica para las cadenas tiene lugar poco antes del nacimiento, de manera que los niveles en el recién nacido son bajos (alrededor del 0,5 %) y alcanzan los valores normales del adulto aproximadamente a los 6 meses de vida.2 El porcentaje de Hb A2 en individuos normales oscila entre 1,8-3,2 %.1
A pesar de que la concentración de esta Hb es baja en sujetos normales, su determinación es muy importante para el diagnóstico de algunos tipos de hemoglobinopatías cuantitativas: la b talasemia heterocigótica, en la cual los niveles de Hb A2 se encuentran por encima del límite superior normal; y la a talasemia ( a -/a a ;a -/ a -;a a /--) con una o 2 cadenas polipeptídicas afectadas en las que la Hb A2 está disminuida.2,3 Esta Hb puede estar aumentada también en la anemia perniciosa, en algunas hemoglobinas inestables y en enfermos que han sido sometidos a trasplante de médula ósea; y disminuida, fundamentalmente en la deficiencia de hierro y la aplasia medular. Recientemente, Perceu y otros demostraron que mutaciones del gen EKFL1 (erythroid Kruppel-like factor) pueden ser responsables también de la presencia de cifras de Hb A2 ligeramente elevadas en ausencia de b talasemia.2-4
Para la cuantificación de la Hb A2 se han utilizado diferentes métodos: electroforesis en acetato de celulosa, en gel de poliacrilamida, cromatografía con microcolumnas, focalización isoeléctrica, cromatografía líquida de intercambio iónico de alta resolución y más recientemente se ha desarrollado la electroforesis capilar. No obstante, la mayoría de ellos son muy laboriosos y la obtención de los resultados es muy lenta.5-8
En este trabajo se presentan los resultados de la cuantificación de la Hb A2 por un método electroforético mediante el empleo de geles de agarosa pre-empacados y el equipo semi-automatizado HYDRASYS 2 de la Firma Sebia (Francia).
MÉTODOS
Se realizó la electroforesis de Hb a 1 942 pacientes atendidos en el Instituto de Hematología e Inmunología (IHI) o remitidos de otros hospitales y servicios de hematología del país, en el período comprendido entre enero y julio de 2011. A 256 casos se les indicó también la cuantificación de Hb A2 por solicitud del médico de asistencia.
La muestra de sangre para el estudio (3 mL) se obtuvo por punción venosa y se recolectó en tubos con anticoagulante (heparina o ácido etilendiamino tetracético-sal disódica; EDTA-Na2). Se lavaron los glóbulos rojos 3 veces con solución salina al 0,9 %. Los glóbulos rojos lavados se conservaron a 4 °C por un período no mayor de 3-5 días. Antes de realizar la electroforesis se preparó un hemolisado con 10 L de glóbulos lavados y 130 L de tris-barbital pH 9,2 0,3.9
Para la corrida electroforética se emplearon geles de agarosa pre-empacados (kit Hydragel 15 hemoglobin(E)) y se realizó en el equipo semi-automatizado Hydrasys 2 de la Firma Sebia (Francia). Para la tinción de las bandas de Hb se utilizó el colorante negro amido.9
La Hb A2 (%) se cuantificó mediante un programa informático diseñado por la compañía Sebia (el equipo de electroforesis está acoplado a una computadora y a un escáner) (tabla).

RESULTADOS
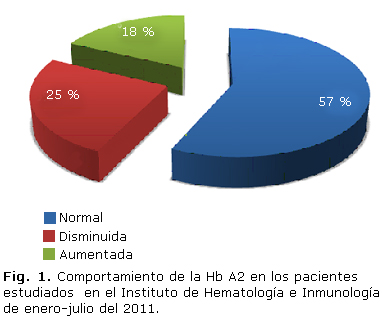
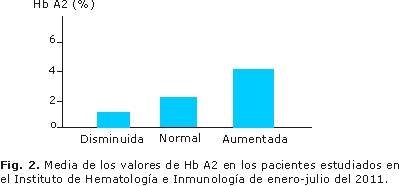
Del total de 256 pacientes, 145 tenían niveles de Hb A2 normal, 65 disminuido y 56 aumentado (Fig. 1). Se calculó la media para cada grupo de pacientes (Fig. 2).
DISCUSIÓN
Las talasemias constituyen un grupo de anemias hemolíticas hereditarias que se caracterizan por la disminución de la síntesis de una o más cadenas de globina. En el momento actual existen más de 250 millones de individuos en el mundo con esta enfermedad.10
La determinación de la Hb A2 es un estudio indispensable en el diagnóstico de las talasemias y de utilidad en otras entidades hematológicas como la deficiencia de hierro y los síndromes de insuficiencia medular.1,10,11
Más de la mitad de los pacientes evaluados presentaron valores normales de Hb A2, lo que sugiere otras causas de anemia en estudio o la presencia de alguna situación especial, como pudiera ser la asociación de una b talasemia con la deficiencia de hierro o con b talasemia en trans, que mantienen la Hb A2 dentro de la normalidad.10,11
Por otra parte, se encontró un mayor número de pacientes con Hb A2 disminuida en relación con la cantidad de pacientes con Hb A2 aumentada. Esto pudiera estar en relación con la frecuencia de las ? talasemias o la deficiencia de hierro en nuestro medio.
En Cuba, en estudios de pesquisa realizados el siglo pasado, se determinó la frecuencia de portadores de b talasemia (0,8 %) por lo que, de acuerdo con la población actual y asumiendo que los matrimonios sean al azar, es de esperar que existan aproximadamente 80 000 individuos con esta enfermedad.12 También se determinó la frecuencia del portador silente de talasemia en individuos no blancos (22,7 %), pero esta no tiene repercusión clínica alguna.
Los resultados demuestran la importancia de continuar perfeccionando el diagnóstico de las talasemias en nuestra población. Con anterioridad, esta técnica se realizaba por cromatografía con resina DEAE-52, la que era muy engorrosa desde el punto de vista técnico, además de la demora en la obtención de los resultados. Con el procedimiento que se emplea actualmente disminuye el tiempo de entrega del informe, además de que se alcanza una mayor sensibilidad y el resultado es más confiable.
REFERENCIAS BIBLIOGRÁFICAS
1. Colombo B, Guerchicoff E, Martínez G. Síndromes talasémicos. Genética y clínica de las hemoglobinas humanas. La Habana: Pueblo y Educación; 1993. p.196-230.
2. Borgna-Pignatti C, Galanello R. Thalassemias and related disorders: quantitative disorders of hemoglobin synthesis. En: Wintrobe's Clinical Hematology. 12th ed. Philadelphia: Lippincott Williams & Wilkins;2009. p. 1084-1119.
3. Kaushansky K, Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Prchal JT. Disorders of globin synthesis: The thalassemias. En: William´s Hematology. 8th ed. New York: McGraw-Hill; 2010.
4. Perceu L, Satta S, Moi P, Demartis FR, Manunza L, Sollanio MC. KLF1 gene mutations cause borderline Hb A2. Blood. 2011;118(6)4454-8.
5. Giambona A, Passarello C, Renda D, Maggio A. The significance of the hemoglobin A2 value in screening for hemoglobinopathies. Clin Biochem. 2009;42:1786-96.
6. Wajcman H, Preahu C, Bardakdjian-Michau J, Prome D, Riou J, Godart C, et al. Abnormal hemoglobins: laboratory methods. Hemoglobin. 2001 May;25(2):169-81.
7. Tangvarasittichai S, Tangvarasittichai O, Jermnim N. Comparison of fast protein liquid chromatography (FPLC) with HPLC, electrophoresis & microcolumn chromatography techniques for the diagnosis of ?-thalassaemia. Indian J Med. 2009;129:242-8.
8. Vrettou C, Kanavakis E, Traeger-Synodinos J, Metaxotou-Mavrommati A, Basiakos I, Maragoudaki E, et al. Molecular studies of thalassemia heterozygotes with raised Hb levels. Hemoglobin. 2000;24:203-20.
9. Hydragel 15 hemoglobin(E). Ref. 4126. Sebia Hydrasys 2 electrophoresis scanning system. Sebia Company. Evry Cedex. France;2010.
10. Villegas A. Talasemias: clasificación y diagnóstico. Haematologica/Edición Española. 2010;95(Extra1)23-32.
11. Bain BJ. Haemoglobinopathy diagnosis: Algorithms, lessons and pitfalls. Blood Rev. 2011;25:205-13.
12. González R, Ballester JM, Estrada M, Lima F, Martínez G, Wade M, et al. A study of the genetical structure of the Cuban population: red cell and serum biochemical markers. Am J Hum Genet. 1976;28:585-96.
Recibido: 15 de agosto del 2012.
Aprobado: 15 de septiembre del 2012.
Lic. Maydelín Miguel-Morales. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800, La Habana, Cuba. Tel (537) 643 8695, 8268, Fax (537) 644 2334. Correo electrónico: rchematologia@infomed.sld.cu
Website: http://www.sld.cu/sitios/ihi
