










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión impresa ISSN 0864-0289
Rev Cubana Hematol Inmunol Hemoter vol.30 no.2 Ciudad de la Habana abr.-jun. 2014
Evaluación de la hemostasia en niños con Síndrome de Ehlers-Danlos tipo III
Hemostasis evaluation in children with type III Ehlers-Danlos syndrome
Dra. Mirta C. Campo Díaz, Dr. Jorge L. Hernández González, Dra. Yumnary Gato Santiesteban, Dr. César Valdés Sojo, Dr. Adalberto Fortún Prieto
Hospital Pediátrico Docente "Pepe Portilla", Pinar del Río, Cuba.
RESUMEN
Introducción: aunque no se ha demostrado la existencia de alteraciones de la hemostasia que formen parte del cuadro clínico del síndrome de Ehlers-Danlos, se han reportado diversas alteraciones de la coagulación en casos aislados, como son eficiencia y alteraciones de la movilidad electroforética de la fibronectina, disfunción de la agregación plaquetaria con prolongación del tiempo de sangramiento, deficiencia de factores VIII, IX, XII y XIII y aumento de la sensibilidad a la aspirina, entre otras.
Objetivo: evaluar la existencia de alteraciones de la hemostasiaen niños con síndrome de Ehlers-Danlos tipo III.
Métodos: se realizó una investigación aplicada, observacional, descriptiva y transversal en 305 niños con síndrome de Ehlers-Danlos tipo III para evaluar, en aquellos con historia de manifestaciones hemorrágicas, la existencia de alteraciones de la hemostasia.Previa suspensión de drogas con acción antiagregante plaquetaria, a todos los pacientes se les realizaron estudios decoagulación y función y agregación plaquetaria.
Resultados: en 181 pacientes se encontró historia de sangramiento espontáneo o traumático, predominantemente cutáneo-mucoso. Elcoagulograma fue normal en todos los casos y el extendido de sangre periférica mostró la presencia de macroplaquetas y escasa formación de grumos como alteración frecuente. Las pruebas de agregación y función plaquetaria evidenciaron la existencia de trastornos cualitativos con predominio de la disminución de la agregación con ADP, sola o combinada con epinefrina y colágeno, y con menor frecuencia trastornos de la disponibilidad de los fosfolípidos plaquetarios. La mayoría de estos pacientes habían utilizado antihistamínicos (ketotifeno) por diversas causas.
Conclusiones: se reporta la presencia de defectos cualitativos plaquetarios en niños con síndrome de Ehlers-Danlos tipo III destacándose el papel de la utilización de drogas antihistamínicas en la aparición de manifestaciones hemorrágicas en estos pacientes.
Palabras clave: síndrome de Ehlers-Danlos tipo III, antihistamínicos, trastornos de la coagulación.
ABSTRACT
Introduction: although the existence of hemostasis disorders as part of type-III Ehlers-Danlos syndrome has not been confirmed, several coagulation alterations have been reported in isolated cases such as: deficiencies and modification in electrophoresis mobility of fibronectin, dysfunction of platelet aggregation with lengthening of bleeding time, deficiency of VIII, IX, XII and XIII factors and increase of aspirin sensitivity, among others.
Objective: evaluate the existence of hemostasis disorders in children with type III Ehlers-Danlos syndrome.
Method: an applied, observational, descriptive and cross-sectional research was carried out in 305 children suffering from type-III Ehlers_Danlos syndrome to evaluate in those having history of hemorrhagic manifestations, the existence of alterations of the hemostasis. Previous suspension of drugs with platelet anti-aggregation action, coagulation and platelet aggregation function studies were carried out.
Results: the study revealed that 181 patients presented history of spontaneous or traumatic bleeding mainly mucous-cutaneous. Coagulogram was normal in all cases and peripheral-blood smears showed the presence of macro-platelets and deficient formation of clots as the most frequent alteration. Aggregation and platelet function tests evidenced the presence of qualitative disorders, where a decrease of aggregation prevailed with the use of adenosine diphosphate (ADP), alone or combined with epinephrine and collagen, and with less frequency, disorders of of platelet phospholipids availability. The majority of these patients presented history of long-lasting use of antihistamines (ketotifen) due to diverse causes.
Conclusions: the occurrence of these qualitative platelet defects in children with EDS-type III is reported, standing out the role of the use of antihistamine drugs on the onset of the hemorrhagic symptoms in these patients.
Keywords: Type-III Ehlers_Danlos syndrome, antihistamines, coagulation disorders.
INTRODUCCIÓN
El síndrome de Ehlers-Danlos es un grupo heterogéneo de trastornos hereditarios del tejido conectivo caracterizado por síntesis anormal de colágeno que afecta la piel, ligamentos, articulaciones y vasos sanguíneos. La anomalía básica en esta entidad es la formación inadecuada del tejido conectivo, que constituye la armazón que da soporte y sostén a la mayoría de los órganos y sistemas.1
En 1997 se realizó la última clasificación del síndrome por un grupo de expertos en Villefrance, que incluye 6 grupos principales, siendo los más frecuentes el I y el III.Cada una de estas variantes posee signos clínicos mayores y menores que definen sus características particulares. La variante III del síndrome debe sospecharse clínicamente y se manifiesta fundamentalmente por hipermovilidad articular generalizada con hiperextensibilidad asociada a anomalías cutáneas de variable intensidad que incluyen hiperelasticidad, piel suave y aterciopelada.2,3
La tendencia hemorrágica en el síndrome de Ehlers-Danlos es más frecuente y severa en el tipo IV, que se considera depende de la fragilidad capilar, adelgazamiento de la piel y ruptura de grandes arterias, más que por defectos en los factores de la coagulación o la función de las plaquetas, 4,5 y hasta el momento, no ha sido demostrada la presencia de alteraciones definidas de la hemostasia que formen parte del cuadro clínico de la enfermedad. Se han reportado alteraciones aisladas de la coagulación, en particular las anomalías plaquetarias en casos aislados del síndrome, que usualmente se han considerado como trastornos asociados a este.6El estudio de función plaquetaria no ha sido evaluado suficientemente para determinar si existen anomalías que participen en las manifestaciones de la entidad. Sin embargo, la tendencia hemorrágica de diverso grado observada en los pacientes con síndrome de Ehlers-Danlos tipo III hace necesaria su evaluación, por lo que se realizó el estudio clínico y de la hemostasia, en particular de la agregación y función plaquetaria, para determinar si existe alguna anomalía que pueda ser considerada parte del cuadro clínico de la enfermedad y los factores que participan en su expresión clínica en esta variante del síndrome.
MÉTODOS
Fueron incluidos 305 pacientes con edades comprendidas entre 5 y 18 años que cumplieron los criterios de síndrome de Ehlers-Danlos tipo III, sin distinción de género, considerados como criterios mayores la presencia de hipermovilidad articular generalizada y afectación cutánea variable, que podrían estar asociados a criterios menores como luxaciones articulares recurrentes, dolor articular crónico o historia familiar sugestiva de la enfermedad. El antecedente de padecer algún trastorno conocido de la hemostasia fue un criterio de exclusión del estudio.
Mediante el interrogatorio de cada paciente o de la persona acompañante se recogió la historia de manifestaciones hemorrágicas y si existió relación con algún elemento desencadenante, como utilización de drogas con acción sobre las plaquetas, fundamentalmente antihistamínicas. Se practicó el examen físico completo con particular énfasis en la evaluación de la movilidad articular según los criterios de Beighton modificados.2
A cada enfermo se le realizóelcoagulogramaen un coagulómetro de 4 canales marca Start de la Diagnóstica Otago, y extendido de sangre periférica. A los que tenían historia de manifestaciones hemorrágicas con resultado de la coagulación normal, se les determinó la disponibilidad de los fosfolípidos plaquetarios según el método de Hardisty, y evaluación de la agregaciónplaquetaria con ADP, epinefrina y colágeno utilizando el agregómetroChorono-log de 4 canales Modelo 490.
Los estudios de agregación y función plaquetaria se realizaron previa suspensión por un período de 30 días de medicamentos con acción antiplaquetaria, incluyendo los antihistamínicos, para evitar resultados falsos positivos.
RESULTADOS
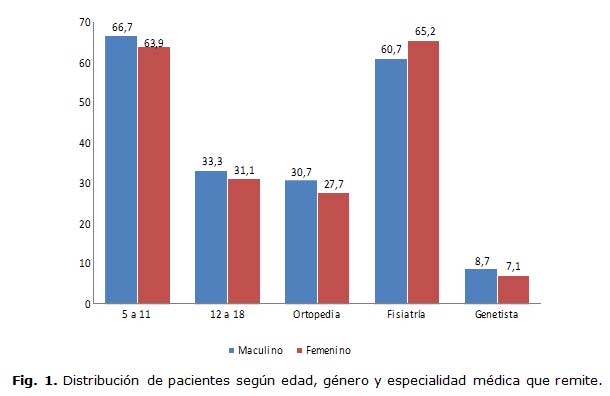
En la figura 1 se muestra la distribución de los pacientes según el género, la edad y la especialidad desde donde fueron remitidos a la consulta de hematología. No se encontró predominio de género y la mayoría de los pacientes tuvieron edades inferiores a 11 años. La especialidad que remitió mayor número de casos fue fisiatría.
Los antecedentes patológicos familiares estuvieron presentes en 223 (73 %) pacientes, predominando la línea materna. Las manifestaciones cutáneas yarticulares fueron las más frecuentes, y se destacó la presencia de dolor articular crónico seguido por la hipermovilidad. La tendencia hemorrágica espontánea o traumática fue poco frecuente.
En la tabla 1 se muestra que la hipermovilidad articular y el compromiso cutáneoestuvieron presentes en todos los niños. El prolapso valvular se encontró en 8 de ellos y fue notable la baja frecuencia de dolor articular crónico.

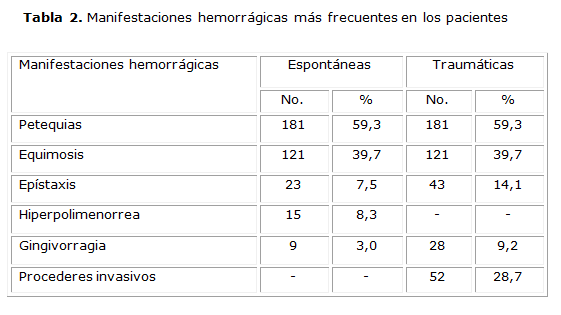
Entre los 181 pacientes con historia de sangramiento (tabla 2) predominaron las manifestaciones hemorrágicas cutáneas (petequias y equímosis). El antecedente de epístaxis y gingivorragia fue predominantemente traumático.
La ingestión de antihistamínicos (ketotifeno) fue encontrada en aproximadamente la mitad de los enfermos con manifestaciones hemorrágicas (91 de 181); y fue excepcional su uso en los que no las presentaron (9 de 124).
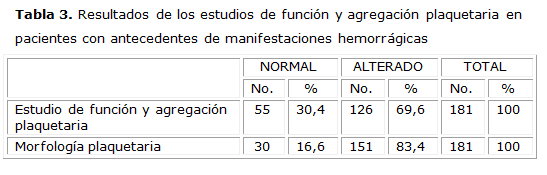
En todos los pacientes evaluadosel estudio de la coagulación fue normal. En los que tenían historia de manifestaciones hemorrágicas se evidenció presencia de macroplaquetas y escasa formación de grumos en el 83,4 % y alteraciones cualitativas plaquetarias en el 69,6 % de ellos, como se muestra en la tabla 3.
En la figura 2 se observa que la disminución de la agregación con ADP fue la alteración más frecuente, sola o combinada. El defecto funcional aislado solo se encontró en 7 pacientes.
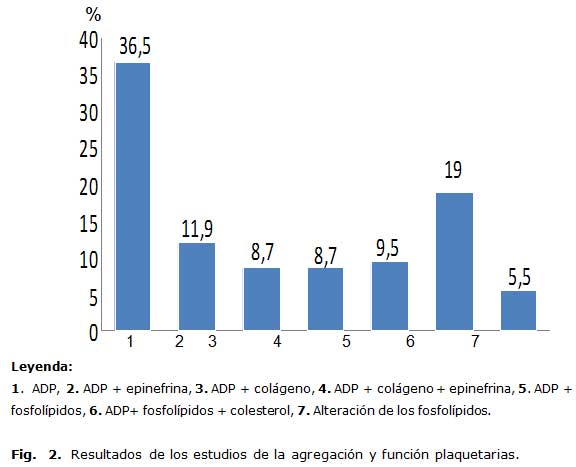
El coagulograma fue normal en la totalidad de los niños estudiados y en la lámina periférica se encontró, con elevada frecuencia, existencia de alteraciones de la morfología plaquetaria. Por su parte, los estudios de agregación y función plaquetaria realizados a los pacientes con tendencia hemorrágica mostraron alteraciones en 126 pacientes, lo que demostró una elevada presencia en esta variante del síndrome, con predominio de disminución de la agregación con ADP, la que fue encontrada en la mayoría de los enfermos (94,5 %), sola o asociada a defectos en la agregación con colágeno o epinefrina, lo que pudiera sugerir un daño en los gránulos plaquetarios por defecto en la liberación de su contenido o en la respuesta plaquetaria a estos agonistas. En los pacientes con historia de sangramiento el 83,4 % mostró alteraciones en la morfología plaquetaria.
DISCUSIÓN
La presencia de diátesis hemorrágica en pacientes con síndrome de Ehlers-Danlos usualmente se ha atribuido a la fragilidad capilar que acompaña a la deficiente estructura del tejido colágeno. Desde hace décadas se han publicado observaciones que indican la posibilidad de que anomalías de la función plaquetaria y otras alteraciones de la coagulación podrían estar presentes en estos pacientes, 7-9 no obstante, dichos resultados no han podido ser utilizados como marcadores propios de este síndrome debido a suasociación infrecuente.
La hipermovilidad articular es común en los niños y ocurre del 8 al 39 % durante la edad escolar. Su prevalencia depende de la edad y el sexo y decrece con la edad.10 Existe debate en la literatura acerca de si la hipermovilidad articular aislada constituye el límite del espectro normal de los movimientos articulares y la mayoría de las personas con este trastorno no presentan problemas derivados de ella.Espor ello que dicho estado ha sido denominado "síndrome de hipermovilidad articular benigna" y forma parte de los trastornos hereditarios del tejido conectivo cuya expresividad clínica es variable. En nuestros pacientes la hipermovilidad articular estuvo presente en todos los niños estudiados, asociada en más de la mitad de ellos a hipotonía muscular. En 52 de ellos se encontró historia de subluxación recidivante y las manifestaciones cutáneas características se encontraron en la totalidad de los enfermos estudiados. Otras alteraciones clínicas propias del síndrome fueron la presencia de genusrecurbatum, escoliosis, miopía y prolapso valvular; lo que marca la diferencia con los niños que presentan hipermovilidad articular "benigna".11
Las manifestaciones hemorrágicas no fueron frecuentes en sus familiares y al igual que lo señalado en la literatura12 en ellos predominó la presencia de sangramiento cutáneo, tanto espontáneo como ante pequeños traumas. Los resultados obtenidosevidencian que la presencia de petequias y equímosis es relevante en los pacientes con síndrome de Ehlers-Danlos tipo III y que los sangramientos mucosos, aunque menos frecuentes, son también un problema clínico que puede estar presente en estos enfermos.
Aproximadamente la mitad de niños con sangramiento cutáneo-mucoso utilizaba de forma continua medicamentos antihistamínicos (ketotifeno), lo que indica que el empleo sistemático de este fármaco puede constituir un factor de riesgo para su aparición.
Los resultados obtenidos ponen de manifiesto la existencia de un trastorno cualitativo plaquetario que pudiera justificar el sangramiento referido, quedando un grupo en que dichas manifestaciones podrían ser secundarias a defectos vasculares de grado ligero que no pueden ser constatados por las pruebas usuales de laboratorio.
El empleo continuado de drogas antihistamínicas (en particular ketotifeno) que inhiben reversiblemente la síntesis de prostaglandinas,13 se encontró en aproximadamente la mitad de los pacientes con historia de trastornos hemorrágicos y solo excepcionalmente en aquellos que no los presentaban, por lo que se recomienda que el usode estos fármacos, muchas veces indiscriminado, debe ser cuidadosamente evaluado debido al riesgo potencial que ello implica a causa de losdefectos hemostáticos señalados.
Estos resultadosaportan un elemento nuevo en el conocimiento de esta variante de la enfermedad. Se describe la existencia de un defecto cualitativo plaquetario por trastorno de la agregación que pudiera estar relacionado con una alteración en el pool de almacenamiento, la utilización o liberación del ADP, solo o combinado con la respuesta inadecuada a otros agonistas, asociado o no a defecto en la exposición de los fosfolípidos plaquetarios, que se encontró con elevada frecuencia en los pacientes con síndrome de Ehlers-Danlos tipo III. Hasta el momento, los trabajos publicados sobre alteraciones cualitativas plaquetarias en la enfermedad sólo se han referido a series pequeñas o reportes de casos, usualmente en otras de las variantes del síndrome. 8, 9
Los hallazgos descritos indican que las alteraciones detalladas podrían ser parte de las manifestaciones de la enfermedad debido a su elevada asociación y además, que la utilización de drogas que aumentan su expresión clínica, como es el caso de los antihistamínicos, pueden hacer evidentes o intensificar estas manifestaciones.
Es necesario incrementar los conocimientos sobre genética molecular y fisiopatología de esta variante del síndrome en la población, frecuentementeno diagnosticado debido a la incorrecta interpretación de los signos y síntomas que lo caracterizan,11 para de esta forma lograr una mejor comprensión y manejo integral de estos niños;unificando los criterios diagnósticos y el tratamiento adecuado desde la atención primaria, donde la detección precoz y la remisión oportuna a los especialistas son vitales para evitar complicaciones, disminuir el daño articular crónico y otras manifestaciones, hasta el nivel secundario de atención donde la evaluación especializada en dependencia de la expresión clínica permitiría el tratamiento adecuado de las diversas manifestaciones del síndrome.
REFERENCIAS BIBLIOGRÁFICAS
1. Ceccolini E, Schwartz R. Ehlers-Danlos Syndrome. 2006. [Citado 12 September 2007].Disponible en: http://emedicine.medscape.com/article/1114004-overview .
2. Beighton P, De Paepe A, Steimann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlossyndrome: revisednosology, Villfranche, 1997.Ehlers-Danlos National Foundation (USA) and Ehlers-DanlosSupport Group (UK). Am J Med Genet. 1998 Apr;77(1):31-7.
3. Levy H. Síndrome de EhlersDanlos tipo hiperlaxitud. Revisión (SED tipo III, Síndorme de EhlersDanlos tipo III. Asociación Síndromes de EhlersDanlos e Hiperlaxitud 2001-2010 [actualizado 2011 Ene 25; citado 2011 Feb 20]. Disponible en: https://sites.google.com/site/rededargentina/sindrome-de-ehlers-danlos-tipo-hiperlaxitud .
4. De Paepe A, Malfait F. Bleeding and bruising in patients with Ehlers-Danlos syndrome and other collagen vascular disorders. Br J Haematol. 2004 Dec;127(5):491-500.
5. Hayward C. Clinical approach to the patient with bleeding or bruising. En: Hoffman: Hematology, basic principles and practice. 6th ed. Philadelphia: Churcill Livingstone Elsevier.2013.p. 1847-56.
6. Onel D, Ulutin S B, Ulutin O N. Platelet defect in a case of Ehlers-Danlos syndrome. ActaHaematol 1973;50:238-44.
7. Beighton P, De Paepe A, Steimann B, Tsipouras P, Wenstrup R J. Ehlers-Danlos syndrome: revised nosology. Villafrance, 1997 Ehlers-Danlos National Foundation (USA). Ehlers.Danlos Support Group (UK). Am J MedGenet 1998;77:31-37.
8. Toneguzzo J, Fourcans G, Gagliardo E, Rodríguez M, Cattaneo M, Izaquirre G, et al. Trastornos plaquetarios. Facultad de Ciencias Médicas. Universidad Nacional de Rosario; Argentina. 2009. [acceso: 4 de junio de 2013]. Disponible en: http://www.clinica-unr.com.ar/Downloads/Revisiones%20-%20Trastornos%20plaquetarios.pdf.
9. Kashiwagi H, Riddle JM, Abraham JP, Frame B. Functional and ultrastructural abnormalities of platelets in Ehlers-Danlos syndrome. Ann Intern Med. 1965;63:249-54.
10. Yenicesu I, Ucan D, Soysal A, Büyükasik Y, Gümrük F. Platelet reliase defect in a child with Ehlers-Danlos syndrome. PediatrHematolOncol. 2000;17:193-4
11. Tofts L J, Elliott E J, Munns C, Pacey V, Sillence D O. The differential diagnosis of children with joint hypermobility: review of the literature. PediatricRheumatol. 2009;7:1-10
12. Campo MC, Fortún Campo A, Beades A, Gato Y, Valdés C. Caracterización del síndrome de Ehlers-Danlos tipo III en Pinar del Río. Rev Ciencias Med (Pinar del Río).2013 May-Jun:17(3);16-24.
13. Díaz M, Ricart G, Escobar A. Mecanismos moleculares de interacción de las plaquetas con el sub-endotrelio vascular: métodos de exploración. En: Hoffman´s Hematology: Basic Principles and Practice, 5th ed. Philadelphia: Churchill LivingstoneElsevier. 2009. 1617-2017.
14. Sharathkumar AA, Shapiro AD. Trastornos de la función plaquetaria. Federación Internacional de Hemofilia. Québec. 2ª. ed. 2008.
Recibido: Septiembre 16, 2013.
Aceptado: Octubre 19, 2013.
CORRESPONDENCIA A:
Dra. Mirta C Campo Díaz. Hospital Pediátrico Docente "Pepe Portilla"Pinar del Río, Cuba.
Email: mcampo@princesa.pri.sld.cu

{kind=link}