










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter vol.31 no.1 Ciudad de la Habana ene.-mar. 2015
PRESENTACIÓN DE CASO
Leucemia mieloide aguda congénita con aplasia del timo
Congenital acute myeloid leukemia with thymic aplasia
Dr. Wilfredo Roque GarcíaI, Dra. Niurka Morán ObregónII, Dra. Maday Rodríguez AcostaII, Dra. Ania Hernández CabezaI, Dra. Anisia Serrano SánchezIII
I Instituto de Hematología e Inmunología, La Habana, Cuba.
II Hospital Ginecobstétrico “Ramón González Coro”, La Habana, Cuba.
III Hospital Pediátrico Docente “William Soler”, La Habana, Cuba.
RESUMEN
La leucemia congénita es una entidad rara que se diagnostica entre el momento del nacimiento y los primeros 30 días de vida. Menos del 1 % de las leucemias de la infancia se diagnostican en el neonato. La hipoplasia severa o aplasia del timo se observa en el síndrome de Di George, que incluye varias malformaciones congénitas y déficit inmunológico, principalmente de células T por hipoplasia del timo, aunque se puede observar aplasia del timo en pacientes sin diagnóstico de Di George. Se presenta un caso diagnosticado como leucemia mieloide aguda congénita. En el momento del nacimiento presentó palidez mucocutánea intensa, petequias, equímosis generalizadas y hepatoesplenomegalia. El diagnóstico se confirmó por la presencia de blastos mieloides en periferia y médula ósea y por la caracterización inmunofenotípica de estas células. La necropsia confirmó la aplasia del timo.
Palabras clave: leucemia congénita, aplasia del timo, síndrome de Di George.
ABSTRACT
Congenital leukemia is a rare disease in which a leukemic process is presented at birth or during the first 30 days of life. Less than 1 % of childhood leukemia is diagnosed in newborns. The severe hypoplasia or total thymic aplasia is seen at Di George syndrome which includes several birth defects and immune deficit, mainly of T cells by thymic hypoplasia; nevertheless, severe thymic hypoplasia can be observed in patients without diagnosis of Di George. We report a case of congenital acute myeloid leukemia who presented intense paleness generalized petechiae and ecchymoses as well as hepatosplenomegaly. The diagnosis was confirmed by the presence of blasts in peripheral blood smear and bone marrow aspirate. Immunophenotyping was performed and contributed to a definitive diagnosis. The autopsy confirmed the thymic aplasia.
Keywords: congenital leukemia, aplasia of thymus, Di George syndrome.
INTRODUCCION
La leucemia congénita es una enfermedad rara que se diagnostica entre el momento del nacimiento y los primeros 30 días de vida. 1 Su incidencia se estima entre uno y cinco por millón de recién nacidos vivos y se reporta que menos del 1 % de las leucemias de la infancia se diagnostican en el neonato.2,5
El timo es el principal órgano de producción, maduración y selección de los linfocitos T. Es relativamente grande (entre 1 000 y 1 500 mm, con un peso promedio de 22 g) y muy activo al nacer. 6 El síndrome de deleción 22q11 se asocia a una variedad de fenotipos clínicos; uno de los más conocidos es el síndrome de Di George que se presenta con malformaciones congénitas asociadas a déficit inmunológico, principalmente de células T por hipoplasia del timo.7 Sin embargo, puede aparecer también hipoplasia o aplasia tímica severa sin que se complete el diagnóstico de síndrome de Di George.
Otro de los diagnósticos con los cuales se debe diferenciar la leucemia congénita es el trastorno mieloproliferativo transitorio (TMT), el cual aparece con frecuencia en los niños con síndrome Down. 8
PRESENTACIÓN DEL CASO
Recién nacida del sexo femenino, hija de una madre de 26 años de edad con historia obstétrica de 11 gestaciones, 9 abortos provocados y 2 partos. El embarazo transcurrió normalmente, con una captación precoz a las 10 semanas, y recibió 12 controles prenatales. No se recogieron antecedentes maternos de consumo de alcohol, tabaco, medicamentos u otras drogas, tratamiento hormonal o radiaciones durante la gestación. No presentó anemia durante el embarazo con una hemoglobina promedio de 111 g/L. La serología (VDRL) del primer y tercer trimestre fue no reactiva y el HIV materno y paterno negativos. Grupo sanguíneo y factor O positivo.
El nacimiento se produjo por cesárea debido a cesárea anterior, a las 40.4 semanas de edad gestacional y no se presentaron complicaciones obstétricas.
La recién nacida pesó 3120 g y tuvo una valoración de Apgar de 5-7, con bradipnea, hipotonía global y frecuencia cardíaca inferior a 100 latidos por min, se aspiró bajo visión laringoscópica y no se obtuvo meconio de la tráquea.
A la exploración física en el momento del nacimiento se observó marcada palidez cutánea con petequias y equimosis generalizadas, tiraje intercostal y abdomen globuloso, de consistencia dura, con hepatomegalia de 4 cm y esplenomegalia de 2 cm por debajo de los rebordes costales respectivos. En la radiografía de tórax se observó gran hepatoesplenomegalia (figura 1).
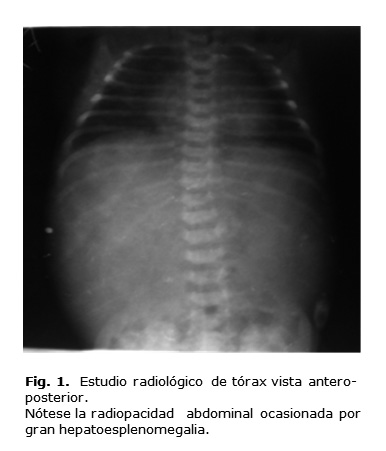
Se trasladó al Servicio de Cuidados Especiales neonatales y se plantearon como posibilidades diagnósticas: la sepsis connatal, infección por citomegalovirus y leucemia congénita aguda.
Se le realizaron los siguientes exámenes complementarios al nacimiento:
Hemograma : hemoglobina 100 g/L, recuento de plaquetas 2 x 109/ L, conteo de leucocitos 150 x 109/L, con un diferencial con predominio de células de aspecto inmaduro.
Glicemia 2.1 mmol/L, grupo y factor O positivo, prueba de Coombs negativa, gasometría capilar que mostró una acidosis metabólica subcompensada. Ionograma normal
Perfil de sepsis : proteína C reactiva (PCR) positiva, lactato 3.7 mmol/L (elevado). Se interpretó como perfil de sepsis positivo con bicitopenia.
Coagulograma : tiempo de protrombina prolongado (control 14 s, paciente 23.8 s), tiempo de tromboplastina activado normal, fibrinógeno 1.7 ( ligeramente disminuido).
Se le realizó cateterización venosa umbilical y vena periférica y se administró transfusión de glóbulos rojos, concentrado de plaquetas, plasma fresco congelado y tratamiento antibiótico con ampicillina y amikacina.
Se tomaron muestras de sangre y orina para estudio de citomegalovirus, que resultó negativo.
A las 27 horas de nacida fue evaluada por los especialistas del Instituto de Hematología e Inmunología, que realizaron frotis de sangre periférica donde se observó leucocitosis y trombocitopenia severas, con predominio de blastos de mediano a gran tamaño (entre 18 - 20 micras), alta relación núcleo-citoplasma, citoplasma ligeramente basófilo, agranular, de núcleo redondo sin escotaduras, cromatina laxa con 1 - 2 nucleolos visibles. En el aspirado de médula ósea no se obtuvo material suficiente para evaluar la celularidad pero se confirma la infiltración por blastos de aspecto mieloide.
El estudio citogenético de la médula ósea mostró 46 XY en 4 metafases y el inmunofenotipaje confirmó la estirpe mielomonocítica al informar CD3 13 %, CD7 11 %, CD13 43 %, CD14 52 %, CD15 8 %, CD22 9 %, CD33 43%, CD41 5%, CD42 16 %.
El tratamiento inicial consistió en exanguinotransfusión y se administró arabinósido de citosina en dosis de 1,3 mg/kg por vía endovenosa, con la que se mantuvo hasta el fallecimiento 7 días después, aunque se logró la reducción de las cifras de leucocitos desde 120 hasta 4.8 x 109/L, y de los blastos, del 82 al 20 %. No se le pudo administrar mercaptopurina ni hidroxiurea ya que tenía suspendida la vía oral debido a sangramiento por el tubo digestivo. Para evitar el síndrome de lisis tumoral se manejó con hidratación amplia y alopurinol en dosis de 10 mg/kg/día.
La paciente tuvo una evolución marcada por sangramiento profuso por los sitios de punturas y el tubo digestivo, lo cual fue manejado con transfusiones de glóbulos rojos y plasma fresco congelado a razón de 10 mL/kg, concentrado de plaquetas, una unidad cada 12 horas y antifibrinolíticos.
A los seis días de vida comenzó con manifestaciones respiratorias, con polipnea, tiraje intercostal y subcostal, crepitantes hacia los vértices y saturaciones de oxígeno entre el 70 y el 83 %. El cuadro empeoró de forma progresiva por lo cual se intentó la intubación endotraqueal a los 8 días de vida, pero no se pudo realizar debido al sangramiento profuso que presentó por el tubo endotraqueal y falleció tras fallar las maniobras de reanimación cardiopulmonar.
La necropsia confirmó que la causa directa de la muerte fue shock hipovolémico, con hemorragias extensas en pulmones, tubo digestivo y vesícula biliar. La médula ósea se encontró hipercelular (80 - 85 %) con hipoplasia trilineal e infiltración difusa por blastos. El análisis inmunohistoquímico fue negativo para CD 3, CD20 y CD79a, y positivo para mieloperoxidasa en la población tumoral (figura 2). Se comprobó la ausencia total de timo y no se encontró malformación de ningún órgano.
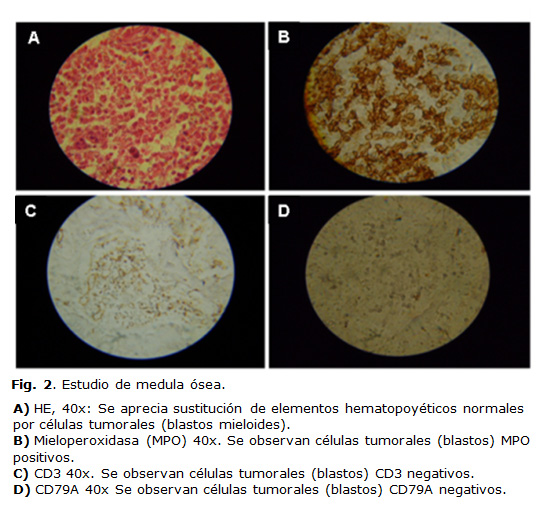
DISCUSIÓN
La leucemia aguda congénita constituye una enfermedad rara cuya etiología no es bien conocida, aunque existen evidencias de que el proceso leucemogénico comienza desde la vida intrauterina. Los criterios diagnósticos que definen esta entidad son: a) presentación al momento del nacimiento y hasta el primer mes de vida; b) incremento de células inmaduras mieloides, linfoides o eritroides; c) infiltración por estas células inmaduras de tejidos no hematopoyéticos; y d) ausencia de otras causas que expliquen esta proliferación celular. 9
La paciente presentó las principales manifestaciones clínicas que definen el síndrome, como palidez cutánea, petequias y equimosis generalizada y hepatoesplenomegalia. La infiltración cutánea o leucemia cutis que se observa en el 30 - 60 % de los casos, 10-12 no se apreció en esta paciente.
El diagnóstico se confirma al estudiar la médula ósea donde generalmente se observa hipercelularidad con una mayor proporción de formas inmaduras, así como ausencia de megacariocitos y de precursores de células del sistema eritropoyético. En este caso no se pudo obtener material del aspirado medular pero la morfología de los blastos observados en el extendido de la médula y en el frotis de sangre periférica fueron compatibles con el diagnóstico de leucemia aguda mieloide y el inmunofenotipaje permitió definir la estirpe mielomonocítica de las células malignas (CD13 43 %, CD14 52 %, CD10 73 %, CD33 43 %).
La autopsia confirmó la ausencia total del timo, lo cual hizo pensar en la posibilidad diagnóstica del síndrome de Di George concomitando con leucemia aguda.
El síndrome de Di George, descrito en 1965, incluye varias malformaciones congénitas y déficit inmunológico, principalmente de células T, por hipoplasia del timo. Existen criterios definitivos, probables y posibles, que diagnostican la entidad; entre los dos primeros es imprescindible que se encuentre reducción de los linfocitos T CD3+ y deleción del cromosoma 22q11.2. Para el diagnostico posible es necesario encontrar aplasia o hipoplasia tímica, reducción de los linfocitos CD3+, hipocalcemia y alguna malformación del sistema cardiovascular. 13
En el caso que se reporta no se realizaron estudios cromosómicos específicos ni recuento de linfocitos CD3, dado el fallecimiento temprano de la paciente y a que la aplasia del timo fue un hallazgo necrópsico. Sin embargo, no se informó malformación cardiovascular ni de ningún otro órgano en el estudio posmortem, lo que reduce la posibilidad diagnóstica del síndrome de Di George.
Otro de los principales diagnósticos con lo cual debe diferenciarse la leucemia congénita es el TMT, anomalía que se observa hasta en el 10 % de los niños con síndrome de Down 8 y que morfológicamente es indistinguible de la leucemia mieloide aguda. Su principal característica es que remite espontáneamente en los primeros meses de vida, por lo que una verdadera diferenciación se hará siempre en retrospectiva. La paciente no presentó ningún estigma físico de síndrome de Down y el estudio citogenético no encontró la trisomía del cromosoma 21 en las metafases estudiadas, por lo que se descartó este diagnóstico.
El pronóstico de la leucemia congénita es malo. En los casos de leucemia mieloide, la sobrevida a los dos años no sobrepasa el 25 % y el índice de recaídas se sitúa en alrededor del 50 %. 3,4
A pesar de que el tratamiento logró la citorreducción y evitó las manifestaciones clínicas y biológicas del síndrome de lisis tumoral, la paciente falleció a los pocos días debido, fundamentalmente, a complicaciones asociadas al sangramiento, sin haberse instaurado el tratamiento específico de la enfermedad hematológica.
En la literatura revisada no se han encontrado reportes de asociación entre leucemia congénita y aplasia del timo, como en el caso presentado.
REFERENCIAS BIBLIOGRÁFICAS
1. van der Linden MH, Creemers S, Pieters R. Diagnosis and management of neonatal leukaemia. Seminars in Fetal & Neonatal Medicine 2012;17(4):192-5.
2. Vormoor J. Leukaemia and cancer in neonatos. Seminars in Fetal & Neonatal Medicine. 2012;17:183-4.
3. González García NE, Broche Cardó R, Trelles Porro L, Cubero Rego MA, Morales Mesa E, Arencibia Núñez A, et al. Leucemia congénita aguda. Rev Cubana Pediatr. 2011 Jun;83(2):193-9.
4. Bresters D, Reus AC, Veerman AJ, van Wering ER, van der Does-van den Berg A, Kaspers GJ. Congenital leukaemia: the Dutch experience and review of the literature. Br J Haematol 2002;117:513-24.
5. Isaacs Jr H. Fetal and neonatal leukemia. J Pediatr Hematol Oncol. 2003;25:348-61.
6. de la Guardia Peña O, Ustariz Garcia C, Garcia García MA, Morera Barrios LM, Chang Monteagudo A, Lavaut Sánchez K. Hipoplasia tímica en un niño con fibrosis quística. Rev Cub Hematol, Inmunol, Hemoter. 2014;30(1):74-80.
7. Lindsay E. Chromosomal microdeletions: dissecting the 22q11 syndrome. Nat Rev Genet. 2001 Nov;2(11):858-68.
8. Pine SR, Guo Q, Yin C, Jayabose S, Druschel CM, Sandoval C. Incidence and clinical implications of GATA1 mutations in newborns with Down syndrome. Blood. 2007;110:2128-31.
9. Wei MC, Dahl GV, Weinstein HJ. Acute myeloid leukemia in children. In: Hoffman R, Benz EJ, Silberstein LE, Heslop HE, Weitz JI, Anastasi J eds. Hematology: basic principles and practice. 6 th ed. Philadelphia: Saunders Elsevier. 2013. p. 913-25.
10. Bas Suarez MP, Lopez BJ, Santana RC, Gresa MM, Diaz PR, Lodos Rojas JC. Congenital acute lymphoblastic leukemia: a two-case report and a review of the literature. Eur J Pediatr. 2011;170:531-4.
11. Benmiloud S, Elhaddou G, Belghiti ZA, Hida M, Bouharrou A. Blueberry muffin syndrome. Pan Afr Med J. 2012;13:23.
12. Nanda A, El-Kamel MF, Al Oneizi EM, Al-Ajmi M, Al Enzei EM, Madda JP. Congenital papulonodular eruption: presenting sign of congenital leukaemia cutis. Clin Exp Dermatol. 2012;37(5):509-11.
13. Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol. 1999 Dec;93(3):190-7.
Recibido: Julio 01, 2014.
Aceptado: Agosto 17, 2014.
Dr. Wilfredo Roque García . Instituto de Hematología e Inmunología. Apartado 8070, La Habana, CP 10800, CUBA. Tel (537) 643 8695, 8268. Fax (537) 644 2334. E mail: rchematologia@infomed.sld.cu

