










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
versión On-line ISSN 1561-2996
Rev Cubana Hematol Inmunol Hemoter vol.31 no.4 Ciudad de la Habana oct.-dic. 2015
ARTÍCULO ORIGINAL
Caracterización clínica y de laboratorio de la anemia hemolítica autoinmune: estudio retrospectivo de 15 casos
Clinical and laboratory features of autoimmune hemolytic anemia: a retrospective study of 15 cases
Dra. Mildrey Gil AgramonteI, Dr. Amel García MonteroI, Dra. Lisette Arias GalánI, Dr. Adrian Romero GonzálezII, Dra. Viviana Cristo PérezI
I Hospital Militar Central “Dr. Carlos J. Finlay”. La Habana, Cuba.
II Hospital Militar Central “Dr. Luis Díaz Soto”. La Habana, Cuba.
RESUMEN
Introducción: la anemia hemolítica autoinmune (AHAI) constituye un cuadro clínico heterogéneo caracterizado por la existencia de autoanticuerpos contra antígenos presentes en la membrana de los eritrocitos del paciente que provocan el acortamiento de su vida media.
Objetivo: conocer las características clínicas y de laboratorio de las anemias hemolíticas autoinmunes diagnosticadas en el centro.
Métodos: se realizó un estudio descriptivo, retrospectivo y de cohorte que incluyó 15 pacientes con el diagnóstico de AHAI en el Hospital Militar Central “Dr. Carlos J. Finlay”, entre enero de 2011 y diciembre de 2013.
Resultados: el rango de edad de los pacientes estudiados fue de 34 a 75 años (mediana de 59 años); de ellos, 8 fueron del sexo femenino y 7 del masculino. El 87 % presentó una AHAI idiopática y el 13 % secundaria. Las secundarias se asociaron con lupus eritematoso sistémico (n = 1) y leucemia linfoide crónica de estirpe B (n = 1). Existió anemia grave de comienzo súbito en el 40 %, e insidioso en el 60 %, íctero en el 73 %, esplenomegalia en el 13 % y dolores articulares difusos en el 20 % de los pacientes. La prueba de Coombs directa resultó positiva en 14 pacientes. Al mes de tratamiento con esteroides, el 33 % presentó una respuesta completa, el 40 % una respuesta parcial y el 27 % no respondió.
Conclusiones: este estudio muestra los hallazgos clínicos y de laboratorio de una pequeña serie de casos adultos con AHAI. La etiología primaria o idiopática fue la más frecuente pero se requiere evolucionar en el tiempo a los pacientes ya que esta entidad puede preceder la aparición de hemopatías malignas o enfermedades del colágeno.
Palabras clave: anemia hemolítica autoinmune, autoanticuerpos.
ABSTRACT
Introduction: autoimmune hemolytic anemia (AIHA) is a heterogeneous clinical picture characterized by the presence of autoantibodies against antigens present on the membrane of the patient's erythrocytes causing shortening of the average life.
Objective: To determine the clinical and laboratory autoimmune hemolytic anemias diagnosed in our hospital.
Methods: adescriptive, retrospective cohort study involving 15 patients with the diagnosis of AIHA was carried out at “Dr. Carlos J. Finlay” Central Military Hospital, between January, 2011 and December, 2013.
Results: the mean age of the patients was 34 - 75 years (median 59 years), 8 were female and 7 male; 87 % had idiopathic AIHA and 13 % secondary AIHA. Secondary hemolytic anemias were associated with systemic lupus erythematosus SLE (n = 1) and B-cell chronic (n = 1) lymphoid leukemia. There was severe anemia (median Hb. 69 g / L) of sudden onset in 40 %, insidious in 60 %, jaundice in 73 %, splenomegaly in 13 % and diffuse joint pain in 20 % of patients. The direct Coombs test was positive in 14 patients. After a month of steroid treatment, 33 % had a complete response, 40 % partial response and 27 % did not respond.
Conclusions: this study shows the clinical and laboratory characteristics of a small number of adults cases with AIHA findings. Primary or idiopathic etiology was the most frequent but evolve evolving patients over time it is required requires patients and that as this entity may precede the onset of hematological malignancies or collagen diseases.
Keywords: autoimmune hemolytic anemia, antibodies.
INTRODUCCIÓN
La anemia hemolítica autoinmune (AHAI) constituye un cuadro clínico heterogéneo caracterizado por la existencia de autoanticuerpos (autoAc) contra antígenos presentes en la membrana de los eritrocitos del paciente, que provocan el acortamiento de su vida media.1 Se distinguen cuatro subtipos según las propiedades específicas del autoAc: el 48 - 70 % son producidas por anticuerpos calientes IgG, reactivos a 37ºC; el 16 - 32 % son producidas por anticuerpos fríos (SAF) IgM, que muestran una actividad óptima a temperaturas menores de 37 ºC (4 - 22ºC); la hemoglobinuria paroxística a frigore (HPF) representa el 5 % en adultos, aunque en los niños aumenta hasta el 32 % y se caracteriza por la presencia de una hemolisina bifásica. La forma mixta representa el 7 - 8 %.1-2
Su incidencia es de 1 a 3 casos por cada 100 000 habitantes/año. En adultos se estima aproximadamente 1:100 000 y en los niños, de 0.2:100 000. 3 En los adolescentes y adultos predomina en el sexo femenino; sin embargo, en la infancia es más frecuente en el sexo masculino. En la edad pediátrica existe un pico de incidencia en los menores de dos años y en los mayores de 12 años.4 Pueden aparecer de forma idiopática entre el 50 % y el 70 %, o ser secundarias a infecciones virales, neoplasias linfoides, enfermedades autoinmunes sistémicas, y con menos frecuencia, asociadas al trasplante de órganos.4-5
El diagnóstico de AHAI se basa en dos aspectos fundamentales: la demostración de la existencia de autoAc y la presencia de un patrón hemolítico (anemia, reticulocitosis, policromatofilia, hiperbilirrubinemia indirecta, aumento de LDH, haptoglobina disminuida y hemoglobinuria).6-7
El tratamiento depende de la evaluación clínica inicial del paciente, las características inmunohematológicas de los anticuerpos y su carácter primario o secundario.8
El pronóstico de las formas secundarias de AHAI radica en la naturaleza de la enfermedad de base. En el caso de las idiopáticas se presenta una evolución crónica con múltiples remisiones y recaídas. Los pacientes usualmente fallecen por fenómenos tromboembólicos o infecciones relacionadas con el tratamiento inmunosupresor, con una mortalidad entre el 36 y el 54 %.9-10
El presente trabajo permitió conocer el comportamiento clínico y de laboratorio de las AHAI diagnosticadas en el centro entre enero del 2011 y diciembre del 2013.
MÉTODOS
Se realizó un estudio descriptivo, retrospectivo y de cohorte que incluyó 15 pacientes diagnosticados como AHAI en el Hospital Militar Central “Dr. Carlos J. Finlay”, con el apoyo del Instituto de Hematología e Inmunología (IHI), entre enero del 2011 y diciembre del 2013.
Se revisaron las historias clínicas y archivos primarios del banco de sangre de dicha institución respetándose la confidencialidad de los resultados individuales.
Se recolectaron los siguientes datos: edad, sexo, hemograma, parámetros clínicos y estudios inmunohematológicos, los que fueron vertidos en una base de datos para su posterior análisis. Se emplearon elementos de la estadística descriptiva: mediana, rango y cálculo de frecuencia.
RESULTADOS
En el periodo analizado se diagnosticaron 15 pacientes con AHAI. De ellos, el 53 % fueron del sexo femenino (n = 8), con una mediana de edad de 59 años (rango 34 - 75 años).
Según la etiología al debut, el 87 % presentó una AHAI idiopática (n = 13) y el 13 % secundaria (n = 2). Las anemias hemolíticas secundarias se asociaron con el lupus eritematoso sistémico (LES) (n = 1) y leucemia linfoide crónica de estirpe B (n = 1).
Como características clínicas, la serie mostró al debut la presencia de anemia grave (mediana de hemoglobina 69 g/L), de comienzo súbito en el 40 % (n = 6) e insidioso en el 60 % (n = 9). Se evidenció la presencia de ictericia en el 73 % (n = 11), esplenomegalia en el 13 % (n = 2) y dolores articulares difusos en el 20 % (n = 3) de los pacientes. Solo en dos casos se constató la presencia de eventos trombóticos, una trombosis venosa profunda femoral y un tromboembolismo pulmonar.
Los resultados de los estudios de laboratorio clínico se muestran en la tabla. El 93.3 % (n = 14) de los casos presentó en el hemograma valores normales de leucocitos y plaquetas. Solo en un paciente se comprobó leucocitosis de 28 x 109 /L con predominio de linfocitos maduros y sombras de Gumprecht en la lámina periférica. El medulograma se realizó a todos los pacientes según lo establecido en el protocolo de AHAI. El 86,7 % (n = 13) se concluyó como una anemia hemolítica crónica, un paciente como una médula ósea reactiva y el otro, compatible con un síndrome linfoproliferativo crónico tipo leucemia linfoide crónica. En este último se corroboró el diagnóstico a través de la biopsia de médula ósea y los marcadores inmunológicos.
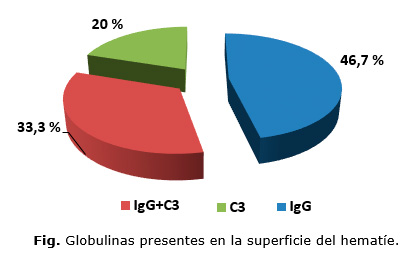
La prueba de antiglobulina directa (PAD) resultó positiva en 14 pacientes (93,3 %). Solo en un caso fue negativa y mediante el estudio inmunoenzimático se demostró la presencia de anticuerpos adheridos al hematíe. La amplitud térmica de los anticuerpos se determinó en todos los casos y el 100 % fue por anticuerpos calientes. Con relación a las globulinas presentes en la superficie del hematíe, su comportamiento se muestra en la figura.
Todos los pacientes requirieron una intervención terapéutica inmediata con esteroides. Solo en dos casos fue necesario el uso de inmunoglobulina G endovenosa. Al mes de tratamiento, el 33 % (n = 5) presentó respuesta completa, el 40 % (n = 6) respuesta parcial y el 27 % (n = 4) no respondió. Estos últimos requirieron el uso de otros inmunosupresores como la ciclofosfamida y la azatioprina, para alcanzar una respuesta parcial.
A los 13 casos de AHAI primaria se les dio seguimiento durante 12 meses para evaluar la historia natural de la enfermedad. Se constató que 2 pacientes evolucionaron a una leucemia linfoide crónica de estirpe B en un periodo de 6 meses y otros 2 a lupus eritematoso sistémico, con una media de 9 meses.
DISCUSIÓN
La AHAI se considera una enfermedad poco frecuente, aunque algunos autores la consideran como la forma más común de anemia hemolítica adquirida11. No se han encontrado diferencias entre grupos étnicos, pero se describe que es más común en el sexo femenino y se incrementa con la edad.12 En la muestra estudiada no existió diferencia significativa con respecto al sexo, lo que pudo estar en relación con el pequeño número de casos; sin embargo, si hubo incremento con la edad.
Desde el punto de vista etiológico, la mayoría de las AHAI son primarias o idiopáticas, observación que se reafirmó en los resultados obtenidos. La AHAI secundaria representa un porcentaje menor, en dependencia de la posibilidad para identificar una enfermedad subyacente. La asociación entre AHAI y la leucemia linfoide crónica de tipo B (LLC-B) está bien documentada y dicha asociación se considera un factor de mal pronóstico en la evolución de la enfermedad.13 Para Comellas et al fue posible evidenciar que el 4 % de los pacientes presentaban síndrome de Evans y un fenómeno antifosfolipídico. Gómez et al reportaron que el 8 % de los pacientes tenían LES al momento del diagnóstico; y otros autores afirman que la AHAI puede ser la primera manifestación de esta enfermedad o preceder su diagnóstico definitivo entre 5 y 10 años.13-14
El inicio de la AHAI suele ser brusco, caracterizado por cifras de hemoglobina que pueden alcanzar valores mínimos de 40 g/L, con repercusión hemodinámica; sin embargo, en esta casuística el mayor número de pacientes tuvo un comienzo insidioso sin repercusión hemodinámica. La destrucción masiva de los eritrocitos ocasiona ictericia y a menudo esplenomegalia.15-16 En el presente estudio, la mediana de hemoglobina fue similar a lo reportado en la literatura, así como las manifestaciones clínicas iniciales.
La AHAI por anticuerpos calientes predominó en todos los pacientes. Según un estudio publicado por el Servicio de Hematología y Hemoterapia del Hospital Universitario Ramón y Cajal de Madrid en el 2009, la AHAI más frecuente fue por este tipo de anticuerpos, que suponen hasta el 70 % de los casos; el SAF el 15,6 %; la HPF el 5 % y las inducidas por drogas el 12,4 %.17
El tratamiento de elección en esta enfermedad al debut son los esteroides y el tratamiento de la enfermedad causal, si existe.18-20 Con los esteroides se puede lograr una remisión del 80 % entre la tercera y cuarta semana de tratamiento.21-25 De no lograrse estabilización de las cifras de hemoglobina existen otras alternativas de tratamiento con fármacos inmunosupresores como la ciclofosfamida, el danazol y la azatioprina.26 La esplenectomía logra neutralizar la efectividad de los anticuerpos IgG, ya que el bazo es el sitio de elección para su destrucción. Otra alternativa es el uso de inmunoglobulina endovenosa en los casos refractarios a la prednisona y a la esplenectomía, pero su efecto es transitorio y solo el 40 % de los enfermos responden. Además, para estos casos refractarios se postula el uso de la terapia biológica como los anticuerpos monoclonales anti-CD 20 (Rituximab) y anti-CD 52 (Alentuzumab). Si existe inestabilidad hemodinámica es necesaria la transfusión de eritrocitos, pero dicha terapéutica debe ser excepcional debido a los efectos secundarios que de ella se derivan.27-29
Como se describe en la literatura, en los pacientes tratados con esteroides se logró una adecuada respuesta hematológica. Solo en un pequeño porciento hubo necesidad de utilizar inmunosupresores y no se utilizó la esplenectomía.
La caracterización clínica y de laboratorio de la muestra diagnosticada con AHAI guarda relación con lo publicado en la literatura, a pesar de ser una pequeña serie de casos. La etiología primaria o idiopática es la más frecuente, pero los pacientes se deben evolucionar, ya que esta entidad puede preceder en el tiempo la aparición de un síndrome linfoproliferativo crónico y enfermedades del colágeno.
REFERENCIAS BIBLIOGRÁFICAS
1. Alfonso ME, Bencomo A. Tratamiento de las anemias hemolíticas autoinmunes. Rev. Cubana Hematol Inmunol Hemoter. 2013;29(4):327-39.
2. Bencomo A, Alfonso ME, Alfonso Y, Salazar MY. Procedimientos para la determinación e identificación de anticuerpos eritrocitarios. Pruebas de compatibilidad pretransfusional. En: Suardíaz J, Cruz C, Colina A: Laboratorio Clínico. La Habana: Ciencias Médicas;2004: p. 575-92.
3. López Martín M. Anemias hemolíticas autoinmunes. Medicina General. 2010;127:186-191.
4. Gupta V, Shukla J, Bhatia BD. Autoimmune hemolytic anemia. Indian J Pediatr. 2008; May;75(5):451-4. doi:10.1007/s12098-008-0071-0.
5. Seung-Woo B, Myung-Won L, Hae-Won R, Kyu-Seop L, Ik-Chan S, Hyo-Jin L. Clinical features and outcomes of autoimmune hemolytic anemia: a retrospective analysis of 32 cases. Korean J Hematol. 2011;46:111-7.
6. Lai M, Leone G, Landolfi R. Autoimmune Hemolytic Anemia with Gel-Based Immunohematology Tests. Am J Clin Pathol. 2013;139:457-63.
7. Aladjidi N, Leverger G, Leblanc T, Quitterie M, Michel G, Bertrand Y. New insights into childhood autoimmune hemolytic anemia: a French national observational study of 265 children. Haematologica. 2011;96(5):655-63. doi:10.3324/haematol.2010.036053.
8. Michel M. Classification and therapeutic approaches in autoimmune hemolytic anemia: an update. Expert Rev Hematol. 2011 Dec;4(6):607-18.
9. Valent P, Lechner K. Diagnosis and treatment of autoimmune haemolytic anaemias in adults: a clinical review. Wien Klin Wochenschr. 2008;120(5-6):136-51.
10. Petz LD, Garraty G. Immune hemolytic anemias. 2nd. ed. Philadelphia: Churchill Livingstone;2004.
11. Petz LD. Diagnostic complexities in autoimmune hemolytic anemias. Transfusion. 2009 Feb;49(2):202-3.
12. Lechner K, Jäger U. How I treat autoimmune hemolytic anemias in adults. Blood. 2010 Sep 16;116(11):1831-8.
13. Rossignol J, Michallet AS, Oberic L, Picard M, Garon A. Rituximab-cyclophosphamide-dexamethasone combination in the management of autoimmune cytopenias associated with chronic lymphocytic leukemia. Leukemia. 2011;25:473-8.
14. Chang A, Chaturvedi Sh, McCrae KR. Thirteen Year Retrospective Analysis of Adult Patients with Autoimmune Hemolytic Anemia at the Cleveland Clinic: Diagnosis and Prevalence of Associated Disorders. Blood. 2013 Nov;122(1):3423.
15. Alfonso Valdés María Elena, Bencomo Hernández Antonio, Espinosa Martínez Edgardo, Guerrero Calderón Raisa, Guerra Borrego Enemilche, Guerrero Calderón Ana Ibis. Caracterización de pacientes adultos con anemia hemolítica autoinmune atendidos en el Instituto de Hematología e Inmunología. Rev Cubana Hematol Inmunol Hemoter [revista en la Internet]. 2009 Dic [citado 2015 Jun 11];25(3). Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892009000300005&lng=es&nrm=iso&tlng=es
16. Naithani R, Agrawal N, Mahapatra M, Kumar R, Pati HP, Choudhry VP. Autoimmune hemolytic anemia in children. Pediatr Hematol Oncol. 2007 Jun;24(4):309-15.
17. Petz LD. Cold antibody autoimmune hemolytic anemias. Blood Rev. 2008 Jan;22(1):1-15.
18. Berentsen S. How I manage cold agglutinin disease. Br J Haematol. 2011 May;153(3):309-17.
19. Barros MM, Blajchman MA, Bordin JO. Warm autoimmune hemolytic anemia: recent progress in understanding the immunobiology and treatment. Transfus Med Rev. 2010;24:195-210.
20. Guilhot F, Roy L, Saulnier PJ, Guilhot J. Interferon in chronic myeloid leukaemia: past and future. Best Pract Res Clin Haematol. 2009 Sep;22(3):315-29.
21. Bencomo-Hernández A, Alfonso-Valdés ME, Munster-Infante A, Basanta-Otero P, Espinosa- Martínez E, Hernández-Ramírez P. Autoanticuerpos en pacientes hematológicos tratados con interferón alfa. Rev Cubana Hematol Inmunol Hemoter. 2000;16(1):49-55.
22. Guía de práctica clínica. Diagnóstico y tratamiento de la anemia hemolítica autoinmune. México: secretaria de Salud;2010. (citado 3 de dic 2012). Disponible en: http://www.cenetec.salud.gob.mx/descargas/gpc/CatalogoMaestro/389_GPC_Diagnostico_y_tratamiento_de_ANEMIA_HEMOLITICA_AUTOINMUNE/GER_ANEMIA_HOMOLITICA_AUTOINMUNE_ADQUIRIDA.pdf .
23. Crowther M, Chan YL, Garbett IK, Lim W, Vickers MA, Crowther MA. Evidence-based focused review of the treatment of idiopathic warm immune hemolytic anemia in adults Blood. 2011 Oct;118(15):4036-40.
24. Gupta N, Sharma S, Seth T, Mishra P, Mahapatra M, Kumar S. Rituximab in steroid refractory autoimmune hemolytic anemia. Indian J Pediatr.2012 Jun;79(6):803-5.
25. Gómez-Almaguer D, Solano-Genesta M, Tarín-Arzaga L, Herrera-Garza JL, Cantú-Rodríguez OG. Low-dose rituximab and alemtuzumab combination therapy for patients with steroid-refractory autoimmune cytopenias. Blood. 2010;116:4783-5.
26. Brauer DL, Edelman B, Rapoport AP, Hess JR, Akpek G. Plasma exchange and rituximab treatment for lenalidomide-associated cold agglutinin disease. Transfusion. 2012 Nov;52(11):2432-5.
27. Kuzmanovic M, Jurisic V. Rituximab for treatment of autoimmune hemolytic anemia. Indian Pediatr. 2012 Aug;49(8):672-4.
28. Fozza C, Longinoti M. Use of Rituximab in autoimmune hemolytic anemia associated with non-hodgkin lymphomas Adv Hematol. 2011;2011:960137.
29. Cooling L, Boxer G, Simon R. Life-Threatening Autoimmune Hemolytic Anemia Treated with Manual Whole Blood Exchange with Rapid Clinical Improvement. J Blood Disorders Transf. 2013;4:163.
Recibido: febrero 19, 2015.
Aceptado: junio 15, 2015.
Dra. Mildrey Gil Agramonte. Instituto de Hematología e Inmunología. Apartado 8070, La Habana, CP 10800, CUBA.
Email: rchematologia@infomed.sld.cu


{kind=link}