










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El cromosoma Filadelfia (Ph por su abreviatura del inglés “Philadelphia) es el resultado de la traslocación balanceada entre los cromosomas 9 y 22, t(9;22)(q34;q11). Se observa en más del 90 % de los pacientes con leucemia mieloide crónica (LMC). 1
Esta alteración cromosómica trae consigo la formación del gen de fusión BCR/ABL1 (Del inglés: break point cluster region - Abelson leucemia) debido a la transposición de la región 3’ del oncogén ABL localizado en 9q34 a la región 5’ del gen BCR en el locus 22q11. El gen de fusión codifica para una proteína quimérica con un dominio tirosina quinasa activado que promueve la continua proliferación celular e inhibe la apoptosis.
La duplicación del cromosoma Ph se adquiere durante la progresión de la enfermedad, aunque puede ser observada durante la fase crónica. (2) Trae como resultado copias adicionales del gen de fusión BCR/ABL1, lo cual se puede observar como un derivativo adicional, isocromosoma del 22 derivativo, entre otras formas de presentación. (3
La terapia con inhibidores de la tirosina quinasa, como el mesilato de imatinib, es usada como tratamiento de primera línea en pacientes con LMC, bloquea la actividad tirosina quinasa e induce la apoptosis. La resistencia a la terapéutica ocurre como resultado del incremento de la expresión del gen de fusión debido a amplificación genómica, mutaciones en el dominio ABL del gen BCR/ABL1 o por alteraciones cromosómicas adicionales, como rearreglos complejos del cromosoma Ph lo cual afecta la interacción con el medicamento. (4,5
Se presentan dos casos con diagnóstico de LMC resistentes al tratamiento con imatinib. A ambos pacientes se les realizó estudio citogenético con bandeo GTG (banda G por tripsina) y Giemsa. En cada caso se observaron 20 metafases y el cariotipo fue descrito acorde al Sistema Internacional para la Nomenclatura de Citogenética Humana 2013.6 El cariotipo (banda G) mostró en ambas muestras, líneas celulares con dos isocromosomas del Ph, 2ider(22)t(9;22), que remplazan al clásico cromosoma Ph. Lo que resulta en la presencia de cuatro copias del gen de fusión BCR/ABL1.
PRESENTACIÓN DE LOS CASOS
Caso 1
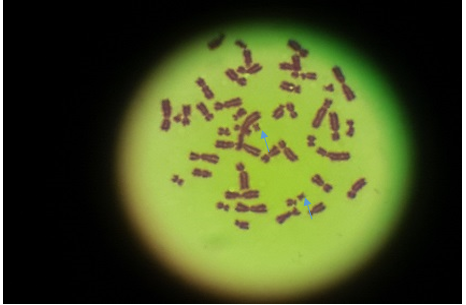
Paciente femenina, de 59 años de edad, con diagnóstico de LMC en el año 2013 que debutó en fase acelerada de la enfermedad, con 8 % de blastos mieloides en médula ósea, esplenomegalia de más de 5 cm, anemia severa y conteo de leucocitos 107x109/L. Llevó tratamiento citorreductor con hidroxiurea y microdosis de arabinósido de citosina. Inició tratamiento con mesilato de imatinib en dosis de 400 mg/d, con remisión hematológica y citogenética de la enfermedad, pero no respuesta molecular. Mantuvo tratamiento por dos años, fecha en que comenzó con pancitopenia y se detectó crisis blástica mieloide de la enfermedad. En el estudio citogenético se observó línea celular con la traslocación t(9,22) y otra con dos isocromosomas del Ph, trisomía13 y un cromosoma marcador (Fig.1). Se aumentó la dosis de imatinib a 800 mg/d. La paciente comenzó con formación de anticuerpos contra el imatinib y se le añadió prednisona en dosis inmunosupresoras, presentó varios ingresos por sepsis urinaria y neumopatía inflamatoria; fue tratada con antibiótico e imatinib (800 mg/d). Falleció al año con un cuadro de neutropenia febril, sepsis grave y crisis blástica mieloide.
Cariotipo: se observó la presencia de dos líneas celulares, la primera con un cromosoma Filadelfia y la segunda con dos isocromosomas del Ph, trisomía13 y un cromosoma marcador (Fig 1) 46,XX,t(9;22)(q32;q11)[15]/49,XX,2ider(22)t(9;22)(q32;q11),+13? +mar [5]

Caso 2
Paciente masculino, de 45 años de edad, con diagnóstico de LMC en fase crónica de la enfermedad desde el año 2001; tratado con Interferón alfa recombinante y que alcanzó control hematológico de la enfermedad. A los cuatro años del diagnóstico, inició tratamiento con mesilato de imatinib en dosis de 400 mg/d, logrando respuesta hematológica y citogenética, pero nunca respuesta molecular. A los siete años de tratamiento con imatinib comenzó con leucocitosis y mielemia, se detectó pérdida de la respuesta citogenética e incremento de blastos mieloides en la biopsia de médula ósea, por lo que se aumentó la dosis de imatinib a 600 mg/d. Tres años después presentó enfermedad diarreica aguda, dolores óseos y se constató disminución de las cifras de hemoglobina y conteo de leucocitos en 53,6 x 109/L; se revaluó y se detectó fase acelerada de la enfermedad. En el medulograma se observó 10 % de blastos mieloides y el cariotipo presentó líneas celulares con dos isocromosomas del Filadelfia (Fig. 2). Se decidió cambiar el tratamiento a nilotinib en dosis de 800 mg/d. Actualmente el paciente que se encuentra en crisis blástica, recibe igual dosificación y esquemas de quimioterapia secuencial con arabinósido de citosina. Pese a esto, no ha tenido buena respuesta y su pronóstico es muy desfavorable. El paciente se encuentra en crisis blástica de la enfermedad con pronóstico muy desfavorable.
Cariotipo: se observó la presencia de dos líneas celulares, la primera con un cromosoma Filadelfia y la segunda con dos isocromosomas del Ph (Fig. 2) 46,XY,t(9;22)( q32;q11)[14] / 47,XY,2ider(22)t(9;22)(q32;q11)[6]
DISCUSIÓN
La presencia de alteraciones cromosómicas adicionales (ACA) en pacientes con LMC se puede interpretar como un signo de evolución clonal y representa un marcador de progresión e inestabilidad cromosómica. Se observan entre el 5-10 % de los pacientes al diagnóstico en fase crónica y entre el 60-80 % de los pacientes con progresión de la enfermedad; por lo que se describe asociación entre los cambios secundarios y la transformación de la enfermedad. (8-10) El primer paciente presentado se encontraba en crisis blástica y el segundo en fase crónica resistente al tratamiento cuando se realizó el diagnóstico de la alteración citogenética.
En los pacientes con LMC y ACA, las alteraciones adicionales en el cromosoma Ph se observan aproximadamente en el 30 %, se pueden observar como un cromosoma 22 derivativo adicional, un isocromosoma del derivativo ider(22)t(9;22) entre otras formas de presentación.5,10) El isocomosoma Filadelfia fue descrito por primera vez por Whang- Peng y posteriormente por otros autores, 11-13) resulta de la fusión de dos cromosomas Ph en la región de los satélites de sus brazos cortos. Se reportó ider(22) formado por la fusión de los brazos largos (q11) del cromosoma Ph en tres pacientes con LMC y en un caso de leucemia linfoblástica aguda.4,14,15) En los dos pacientes presentados se observaron en el cariotipo líneas celulares con 2ider(22)t(9;22) fusionado en sus brazos cortos, lo que trae consigo la presencia de cuatro genes de fusión BCR/ABL1. Además, en la primera paciente se observó en las metafases con isocromosomas, un cromosoma marcador y un cromosoma adicional que por su morfología impresionó pertenecer al grupo D (cromosomas 13, 14 o 15), pero su identificación resultó difícil debido a que el bandeo cromosómico no mostró la mejor calidad.
La ganancia del cromosoma Ph se cree que sea resultado de una no disyunción mitótica o de un error en la segregación, aunque no se conoce la causa con exactitud. Lo que sí es conocido, es que la presencia de esta alteración genética se asocia a inestabilidad genómica, peor pronóstico y mayor probabilidad de resistencia a la terapia con inhibidores de tirosina quinasa. 5,11,16
Estas alteraciones pueden conferir proliferación independiente de BCR/ABL1 y disminución de la sensibilidad al medicamento. Lo que contribuye a la expansión de la población celular Ph +, se acentuá la capacidad de proliferación, la resistencia a la apoptosis y la pérdida de la capacidad de diferenciación.17,18) La paciente falleció y el otro caso se encuentra actualmente en crisis blástica con pronóstico muy desfavorable.
El estudio citogenético por técnica de banda G permitió la identificaciónde alteraciones cromosómicas, lo que facilita una mejor caracterización genética de los pacientes para evaluar el pronóstico y la respuesta al tratamiento.

