










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La drepanocitosis es la anemia hemolítica intracorpuscular determinada genéticamente, más frecuente en el mundo. Se debe a una mutación en la posición 6 del gen que codifica para la cadena ( de la globina, en la que el ácido glutámico es sustituido por la valina.1
Su frecuencia es mayor en África, el Medio Oriente, sur de Italia, norte de Grecia, sur de Turquía, provincias occidentales de Arabia Saudita y la India. Fue trasladada por el comercio de esclavos a Estados Unidos de Norteamérica, América Central, el Caribe y algunos países de América del Sur.1
La forma más frecuente de drepanocitosis en Cuba, es la anemia drepanocítica (AD) o hemoglobinopatía SS, le siguen en frecuencia la hemoglobinopatía SC (HSC) y la S( talasemia (S( tal).1
La drepanocitosis constituye un grupo de anemias hemolíticas crónicas, que se caracterizan todas, por crisis vasoclusivas, anemia hemolítica, vasculopatía, lesiones orgánicas diseminadas agudas y crónicas y una mortalidad prematura.1
Dentro de las complicaciones observadas de manera frecuente en estos pacientes se encuentran, las crisis aplásicas, que son producidas por una infección por la cepa B19 del parvovirus humano (PVH B19), que determina una supresión de la eritropoyesis durante 7 a 10 días. Debido a que la vida media del eritrocito en la drepanocitosis está muy acortada, ocurre un descenso de la cifra de hemoglobina en pocos días. El diagnóstico se realiza porque existe: reticulocitopenia y en el medulograma se observa la hipoplasia del sistema eritropoyético.1-3
Se sabe que la cepa de PVHB19 puede ser causante de anemias hemolíticas autoinmunes (AHAI) en los pacientes previamente infectados por el mismo.1,2
Las causas más frecuentes de las anemias hemolíticas extracorpusculares suelen ser los problemas mecánicos, químicos, infecciosos o inmunológicos; entre estos últimos se encuentran las AHAI, las cuales, desde que Coombs las describió en 1945 e introdujo la prueba que lleva su nombre, fueron reconocidas como tales en el contexto biomédico.4
Esta prueba es fundamental para el diagnóstico de la hemólisis por procesos inmunes, pues en ella se demuestra la presencia de autoanticuerpos o elementos del complemento unidos a la superficie del hematíe (Coombs directo) o el incremento en el suero de anticuerpos (Coombs indirecto). Se desconoce el mecanismo por el cual, el organismo forma anticuerpos contra sus propios glóbulos rojos, pero sí se conoce los mecanismos inmunológicos capaces de destruirlos.4
Hasta donde se conoce, la asociación entre la drepanocitosis y la AHAI es poco frecuente; solo se han descrito unos pocos casos de infección aguda por PVH B19 como causa de AHAI, por formación de anticuerpos dirigidos contra los glóbulos rojos. Dada la poca frecuencia de esta asociación se presenta el caso de una paciente con antecedentes de hemoglobinopatía SC que posterior a una crisis aplásica presentó una AHAI.
PRESENTACIÓN DEL CASO
Paciente femenina, se color de piel negra, de 66 años de edad, con antecedentes de hemoglobinopatía SC e historia hematológica de crisis relacionadas con síndrome torácico agudo, hepática a repetición y vasoclusivas dolorosas frecuentes; con requerimientos de transfusiones en varias ocasiones, sin demostración de aloanticuerpos y sin utilización de hidroxiurea como tratamiento, cuya hemoglobina habitual oscila entre 110 - 115 g/L.
En junio del 2016, asistió al servicio de urgencia por astenia marcada, palidez y toma del estado general. Al examen físico se detectó palidez cutánea mucosa y taquicardia
En los exámenes complementarios se observó: hemoglobina de 66 g/L, disminuida en relación a los valores habitualmente tolerados por la paciente; conteo de reticulocitos de 0.1 %, disminuido, leucocitos y conteo de plaquetas en valores normales. Se le realizó el aspirado de médula ósea y se comprobó la presencia de una depresión de la serie eritropoyética con cambios megaloblásticos (Fig. 1).
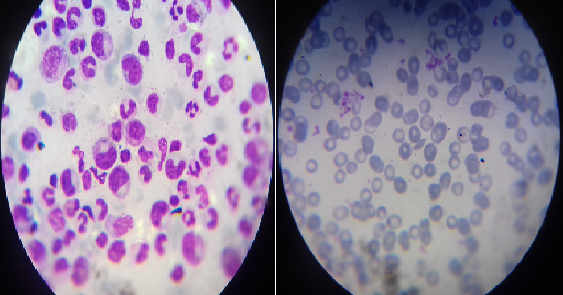
Fig. 1- Hipoplasia del sistema eritropoyético en paciente con hemoglobinopatía SC y anemia hemolítica autoinmune (AHAI) secundaria a infección por parvovirus humano B19.
Se le diagnosticó una crisis aplásica y se ingresó en el servicio de Adultos, donde se transfundió con concentrado de eritrocitos. Alcanzó la recuperación clínica y hematológica en un término de 10 días. Se le repitió el aspirado de médula ósea y se encontró mayor integridad del sistema eritropoyético, fue egresada y se continuó su seguimiento por consulta externa.
En agosto de 2016, con solo 2 meses de haber presentado el evento anterior, acudió al servicio de urgencia con astenia marcada y palidez cutáneo mucosa intensa, disminución de las cifras de hemoglobina en relación con los valores basales de la paciente (60g/L), reticulopenia. En el estudio se encontró una prueba de Coombs directa (PAD) positiva; el reactivo monoespecífico anti-Ig G negativo y con el reactivo monoespecífico anti-C3d positivo, eluído negativo, una prueba de Coombs indirecta (PAI) negativa y con título de crioaglutininas Ig M > 1/64. En el aspirado de médula ósea se constató hipoplasia de la serie eritropoyética y estudios de PAD positivo a C3d, PAI negativo.
Se le realizaron estudios de anticuerpos antinucleares, anti-ADN, factor reumatoideo, cuantificación de inmunoglobulinas, estudios virológicos (hepatitis B, C y HIV) que fueron negativos; así como una prueba serológica para virus de Epstein Barr y citomegalovirus que mostró una infección pasada. Se ingresó y se transfundió con concentrado de eritrocitos precalentados.
Con los resultados obtenidos se diagnosticó una AHAI, pero resultaba llamativo que la paciente tenía una reticulocitopenia, cuando en la AHAI existe una reticulocitosis- Se demostró la presencia de autoanticuerpos fríos, con un C3d positivo en médula ósea, elemento de gran valor que explicaba la existencia de una reticulocitopenia por la destrucción de los progenitores eritroides por los autoanticuerpos.
Se comenzó tratamiento con prednisona en dosis inmunosupresoras de 1 mg/kg/d teniendo en cuenta la edad de la paciente, se usaron 60 mg diarios. Al término de 7 días se le repitió hemoglobina con un incremento de 1 g en relación a las cifras anteriores; se continuó con esta dosis durante 1 mes, las cifras de hemoglobina continuaron ascendiendo y se comenzó a disminuir paulatinamente el esteroide hasta que sin necesidad de su uso se mantuvo estable, con sus cifras de hemoglobina habituales y sin complicaciones. Se continuó el seguimiento en consulta especializada, sin complicaciones hasta el momento de elaborar este reporte.
DISCUSIÓN
La infección por PVH B19 es la causa de la mayor parte de los casos de crisis aplásica transitoria, que aparecen de forma busca en pacientes con enfermedades hemolíticas crónicas, como es el caso de la drepanocitosis.2,3 Sin embargo, la asociación entre drepanocitosis y AHAI es poco frecuente. 1-3
Se han descrito unos pocos casos de infección aguda por PVHB19 como causa de AHAI, por medio de la formación de anticuerpos dirigidos contra los glóbulos rojos. 2-4) El mecanismo específico de producción de AHAI por PVHB19 permanece sin esclarecer, pero es importante subrayar que la presencia de una infección aguda por PVHB19 debe sospecharse ante un episodio de AHAI que curse con reticulopenia e hipoplasia de los precursores eritroides de la médula ósea. 2-4)
En la literatura se recogen reportes de casos de AHAI por crioaglutininas secundaria a infección por Ebstein Barr, citomegalovirus, hepatitis viral A, B, C y VIH. 5
En este caso en particular se presentó el evento primario de la crisis aplásica y posteriormente apareció la AHAI que tuvo una respuesta favorable al tratamiento esteroideo impuesto dada la posible naturaleza autoinmune de la AHAI. Aunque el uso de los esteroides es controvertido, estos resultados se encuentran acorde con lo revisado por la literatura nacional e internacional.1,5,6
La evolución favorable observada permite afirmar que el tratamiento con esteroides es efectivo en AHAI independientemente de la causa que la produzca.

