










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La terapéutica en los síndromes mielodisplásicos (SMD) evoluciona a la par de los nuevos descubrimientos y hacia ellos se enfocan las propuestas de los nuevos agentes terapéuticos. Dos moduladores epigenéticos, los agentes hipometilantes (AHM), la azacitidina y la decitabina y un inmunomodulador, la lenalidomida, fueron aprobados por la FDA (siglas del inglés Food Drug American) y considerados como tratamiento de primera línea hace más de una década. Diferentes estudios muestran los resultados satisfactorios obtenidos en alrededor del 50 % de los casos tratados, con baja toxicidad e influencia en la calidad de vida de los pacientes.1,2,3,4,5,6
A pesar de que se obtuvieron resultados similares con ambos AHM, solo la azacitidina ha demostrado tener influencia en la supervivencia, ya sea con remisiones completas o parciales. Sin embargo, un porcentaje no despreciable de pacientes tratados con los AHM no logran ningún tipo de respuesta o la enfermedad progresa rápidamente después de 6 ciclos de tratamiento (resistencia primaria), y en otros la duración de la respuesta inicial es menor de 6 meses (resistencia secundaria). Diversos estudios indican que con estos agentes no se erradica el clon leucémico. 7,8,9,10
Debido al elevado número de pacientes que no responden a los AHM, constantemente se realizan estudios en la búsqueda de las causas asociadas al fallo. Nazha y otros publicaron algunos factores predictores de respuesta a los AHM, con una mayor significación estadística para la edad (mayores de 75 años), la dependencia de transfusiones de glóbulos rojos, el conteo de plaquetas menor de 30 x 109/L y la presencia de alteraciones citogenéticas de mal pronóstico, fundamentalmente las alteraciones complejas. En ese artículo, también se señalan como predictores negativos el ECOG (siglas del inglés Eastern Cooperative Oncology Group) mayor de 1 y más de 20 % de blastos en médula ósea.10
Variadas estrategias terapéuticas se estudian hoy en ensayos clínicos para su uso como monoterapia o combinado con otras ya conocidas. La mayor parte de las estrategias están enfocadas en las principales alteraciones genéticas encontradas hasta hoy en los SMD, que ya suman mutaciones en más de 40 genes y están presentes en más 80-90 % de estos enfermos.11,12) En esta revisión se van a analizar las opciones actuales para el tratamiento de los síndromes mielodisplásicos.
Métodos
Se realizó una revisión de la literatura a través del sitio web PubMed y el motor de búsqueda Google académico de artículos publicados en los últimos 5 años. Se emplearon como palabras clave: síndromes mielodisplásicos, agentes hipometilantes, nuevas opciones terapéuticas. El 74,2 % de los trabajos seleccionados fueron artículos originales y de revisión publicados entre 2015-2019 en los idiomas inglés y español, un 14,2 % fueron ensayos clínicos actuales y presentaciones en eventos internacionales. El 11,4 % de la bibliografía correspondió a 4 años anteriores. Se hizo un análisis y resumen de la bibliografía; se tomaron los aspectos más importantes referidos al tema.
Análisis y síntesis de la información
La mayor parte de los tratamientos que se ensayan en la actualidad están dirigidos contra los eventos epigenéticos fundamentales: la hipermetilación, las modificaciones de las histonas diacetilasa, la regulación epigenética y la activación de la respuesta inmune citotóxica contra clones anormales, entre otras. En la tabla 1 se muestran algunos de los medicamentos en diferentes fases de ensayos clínicos.
Tabla 1 Algunos tratamientos dirigidos contra eventos epigenéticos en pacientes con síndromes mielodisplásicos
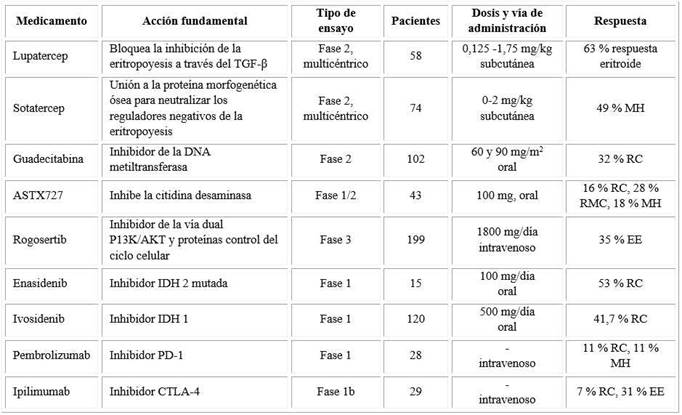
RC: remisión completa MH: mejoría hematológica EE: enfermedad estable, RMC respuesta medular completa, TG F-β factor de crecimiento transformante β, IDH isocitrato dehidrogenasa, PD-1 ligando de la muerte celular programada, CTLA-4 antígeno de linfocitos T citotóxico- 4
Síndromes mielodisplásicos de bajo riesgo
El uso de la Lenalidomida, y en su defecto la Talidomida, en pacientes con SMD de riesgo bajo e intermedio, especialmente en pacientes con el subtipo 5q-, en que además del efecto inmunomodulador se plantea tiene efecto antinflamatorio y antiangiogénico, continua siendo una opción válida.1
Por otra parte, los estimulantes de la eritropoyesis, los miméticos de la trombopoyetina (eltrombopag y romiplostin) y la combinación de eritropoyetina en dosis de 30 000 a 60 000 unidades con factor estimulante del crecimiento granulocítico, mantienen su indicación como tratamiento de soporte. Los AHM también son de utilidad en este tipo de pacientes.13,14
En la actualidad se considera que todo paciente con SMD debe ser candidato a alguna terapia, fundamentalmente si presenta mutaciones específicas, entre ellas IDH 1/2, SRSF2, SF3B1 y U2AF1, que permitan incluirlos en algún ensayo clínico. Entre los ensayos experimentales se preconizan los inhibidores de citocinas, Luspatercept y Sotatercept, un nuevo AHM (Guadecitabina) y los inhibidores del espliceosoma.15
Inhibidores de citocinas
Luspatercept: Es una proteína de fusión recombinante que contiene el receptor de la activina tipo II B modificado, unido a la fracción Fc de la Inmunoglobulina G 1 humana.15,16 Su acción es bloquear los inhibidores de la eritropoyesis, específicamente el factor β transformador del crecimiento, conocido por sus siglas del inglés TGF-β. Este al unirse al factor de diferenciación de crecimiento 11, conocido también como proteína morfogenética ósea y a otros ligandos de la superfamilia TGF-β, promueve la diferenciación eritroide en una etapa tardía.
En un estudio multicéntrico fase II realizado en Alemania,16 el Luspatercept fue efectivo en el tratamiento de la anemia en los SMD de bajo riesgo con el uso de dosis escalonadas de 0.125 a 1.75 mg/kg administradas por vía subcutánea (SC). La respuesta fue mejor en pacientes con más de 15 % de sideroblastos anillados y la mutación SF3B1. Un dato importante es que su acción no tiene relación con los niveles de eritropoyetina endógena, como sucede con otros estimuladores de la eritropoyesis.
Sotatercept: Es una proteína de fusión recombinante del receptor de la activina tipo II-A que actúa como un ligando para neutralizar el regulador negativo de la eritropoyesis en etapa tardía.17
Un estudio multicéntrico fase II en Estados Unidos y Francia publicó los resultados de este tratamiento en pacientes de riesgo bajo e intermedio 1, de acuerdo al sistema internacional de puntaje pronóstico IPSS (siglas del inglés International Prognostic Scoring System), que tenían anemia y requerimientos transfusionales sin respuesta a estimuladores de la eritropoyesis. Los pacientes con síndrome 5 q- solo se incluyeron si existía fallo documentado del tratamiento con Lenalidomida. Se estudiaron varios grupos de pacientes con dosis de Sotatercept de 0,1 a 2 mg/kg por vía subcutánea cada 3 semanas hasta 5 dosis. Se reporta respuesta eritroide en el 63 % de los pacientes incluidos e independencia de transfusiones en el 38 % de los casos tratados, con buena tolerancia al tratamiento.
Síndromes mielodisplásicos de alto riesgo
Posterior al fracaso con los AHM, las opciones en estos pacientes son muy escasas si no son candidatos a trasplante de médula ósea alogénico. La quimioterapia intensiva similar a la usada en el tratamiento de la leucemia mieloide aguda con esquemas 3/7 en dosis intermedias y altas de citarabina, así como las bajas dosis de citarabina no han mostrado ventajas.14,15 Por tanto, en función de los diferentes adelantos en la biología de la enfermedad, se impone la necesidad de desarrollar nuevos medicamentos. Así se destacan nuevos hipometilantes, inhibidores de la isocitrato deshidrogenasa 1 y 2, inhibidores del espliosoma e inhibidores multiquinasas, entre otros.15
Hipometilantes
Después de los resultados alcanzados con la azacitidina y la decitabina, nuevos AHM están en investigación, algunos de ellos de uso oral.
El fallo o resistencia a los AHM se relaciona con alteraciones en el transporte y metabolismo de los AHM conocidos (azacitidina y decitabina), específicamente el incremento de la actividad de la enzima citidina desaminasa. Esto pudiera resultar de resistencia a estos agentes debido a la incorporación deficiente al ADN/RNA celular y por tanto provocar fallo en el tratamiento.18
La Guadecitabina, un AHM de segunda generación, es un dinucleótido de la decitabina, resistente a la degradación por la citidina desaminasa, por lo que puede inhibir de forma potente la actividad de la metiltransferasa. Tiene una vida media mayor que los otros AHM y la enzima se libera de forma gradual y progresiva.
Se empleó en pacientes con SMD de riesgo intermedio y alto sin antecedentes de tratamiento y refractarios a la azacitidina. Se realizaron 2 grupos de dosis (60 y 90 mg/m2 por vía subcutánea por 5 días cada 28 días) en los que incluían en ambos pacientes de novo y refractarios o recidivantes. La respuesta fue favorable en el 55 % (25 de 49) de los casos sin tratamiento previo y en el 51 % (23 de 53) de los refractarios o recidivantes en ambos brazos del estudio. No existió diferencia significativa en los resultados en ambos grupos de dosis por lo que recomiendan la dosis de 60 mg/m2. Hubo 7 fallecidos, 6 de ellos refractarios con efectos adversos grado 3 (trombocitopenia, neutropenia, anemia y neumonía).18
El ASTX727 es una combinación de decitabina oral con Cedazuridina, (35 mg de decitabina/100 mg de cedazuridina). Este último es un inhibidor de la citidina desaminasa que pudiera mejorar la biodisponibilidad de la decitabina. En la reunión anual de la Sociedad Americana de Hematología (ASH, siglas en inglés) del año 2017, se presentaron los resultados de un estudio fase 2 en pacientes con SMD de riesgo intermedio 1 y 2 y alto y los resultados fueron satisfactorios en el 62 % de los enfermos tratados y ocurrieron eventos adversos en el 25 % de los casos, dados fundamentalmente por citopenias y fatiga.19
Inhibidores de la isocitratodeshidogenasa 1y 2
Normalmente, la familia de las isocitrato deshidrogenasa (IDH) convierte la isocitrato a α-cetoglutarato por decarboxilación oxidativa, en la mitocondria. Las mutaciones de esas enzimas causan conversión de este metabolito en 2-hidroxiglutarato, un oncometabolito que en altas concentraciones altera la metilación del ADN y bloquea la diferenciación de las células progenitoras hematopoyéticas. Las mutaciones IDH 1 y 2 tienen una baja frecuencia en los SMD (4-12 %) y se asocian a la edad avanzada y la trombocitosis; además, se vinculan a otras mutaciones como la DNMT3A, ASXL1 y SRSF2. Su valor pronóstico es controversial, pero se les atribuye un impacto negativo, especialmente en los pacientes de bajo riesgo.5,20
Algunos agentes inhibidores de la IDH han sido usados solos, en combinación con AHM o con quimioterapia intensiva con resultados alentadores.
Enasidenib: es un potente inhibidor de la enzima IDH 2 mutada, de uso oral, está aprobado por la FDA para el tratamiento de la leucemia mieloide aguda.20
En un estudio de Stein y colaboradores en pacientes con SMD, la mutación estuvo presente en el 5 % de los sujetos, la mitad de los casos incluidos eran de alto riesgo y la dosis empleada fue de 100 mg/día por vía oral en ciclos de 28 días. En esta serie se informaron respuestas hematológicas en el 53 % de los pacientes con buena tolerancia.21)
Ivosidenib: es un inhibidor de la IDH 1, también de uso oral. En la reunión anual de la ASH del año 2018 se presentó un trabajo en el que se analizaron por separado 12 pacientes con SMD incluidos en un ensayo clínico con este medicamento y se reportaron 5 remisiones completas (41,7 %); de ellos, 1 con aclaramiento de la mutación y 9 con independencia de transfusiones de más de 50 días.22
Inhibidores multiquinasas
El Rigosertib es una bencil estiril sulfona sintética que bloquea la señalización del oncogen RAS. Este fármaco bloquea la activación de la vía fosfatidil inositol 3 kinasa/ polo-like kinasa (PI3K/AKT) y las proteínas de control del ciclo celular. En diferentes estudios ha mostrado reducir el número de blastos en médula ósea en pacientes con SMD.15
García Manero y otros publicaron los resultados de un estudio controlado fase III con Rigosertib contra el mejor tratamiento de soporte en pacientes de alto riesgo con refractariedad al tratamiento con AHM con una mediana de seguimiento de 19,5 meses. La dosis de Rigosertib fue de 1800 mg/día en infusión continua por 72 h en semanas alternas por 8 ciclos. Los investigadores plantean ventajas en la sobrevida global para los pacientes menores de 80 años y con resistencia primaria a los AHM. En este estudio la respuesta a los parámetros periféricos fue mínima, no reportaron respuestas completas, ni parciales, pero refirieron remisión medular completa en el 20 % de los pacientes tratados y enfermedad estable en el 35 % de ellos.23
Otros ensayos clínicos están en marcha con el uso de Rigosertib oral como monoterapia en pacientes de riesgo bajo e intermedio 1 (NCT01584531 y NCT01904682) o combinado con azacitidina (NCT01926587).24,25,26
Otros inhibidores multiquinasas como Volasertib inhibidor PLK-1 (polo-like kinasa) (NCT01957644), Binimetinb MEK, inhibidor RAS de uso oral y un Pan-PIM quinasa inhibidor oral (LGH447 NCT02078609) están también en investigación.
Otras líneas de tratamiento
Inhibidores del BCL 2
La vía intrínseca de la apoptosis celular está controlada por la familia BCL2 a través de la activación de las caspasas 3 y 7 y la sobrexpresión de esta familia con la correspondiente desregulación en la apoptosis se ha demostrado en los SMD.27
Venetoclax: es un potente inhibidor selectivo BCL2, de uso oral, que tiene actividad antiapoptótica in vitro en células de pacientes con SMD de alto riesgo, se utilizó en un protocolo que incluyó un pequeño número de pacientes con SMD refractarios o en recaída combinado con AHM o bajas dosis de citarabina. La dosis media que se administró fue 300 mg y concluyeron que era una opción viable como terapia de salvamento, particularmente en pacientes con diploidia o riesgo intermedio.28
Trióxido de arsénico: tiene actividad proapóptótica, antiproliferativa, antiangiogénica y actúa sobre la desregulación de la familia BCL-2. Su uso solo o combinado con otros agentes en el tratamiento de los SMD mostró resultados satisfactorios en un porcentaje de los casos tratados.29
La asociación del arsénico con Talidomida fue efectiva y bien tolerada en pacientes con SMD, sobre todo en aquellos con inv3 (3q 21; 3q26).30
Inhibidores de la histona diacetilasa (IHD)
Las histonas pueden ser modificadas por diferentes procesos bioquímicos, pero la acetilación es una de las principales modificaciones asociadas a la expresión de genes y esta es controlada por las enzimas acetiltransferasa y diacetilasa. Específicamente, la histona diacetilasa actúa modificando la expresión génica para restaurar la diferenciación y la muerte celular programada.
Existen 5 clases (I-IIA-IIB, III y IV) con diferentes mecanismos de acción como remodelación de la cromatina, inhibición de la angiogénesis, estimulación de la apoptosis, producción de radicales libres, interferencia en las funciones de las proteínas chaperonas y desregulación del ciclo celular.31
En general, los IHD no han demostrado gran eficacia como único tratamiento, pero asociados con otros medicamentos (AHM, idarubicina, citarabina, bortezomib y ácido trans-retinoico, entre otros) han mostrado resultados prometedores.
Entre los IHD se destacan estudios con Vorinostat, Panobinostat, Etinostat y Romidepsin. También en este grupo está al antiguo valproato de sodio, conocido antiepileptógeno, que se ha reportado disminuye la proliferación e induce la diferenciación de blastos mieloides en pacientes con SMD y LMA.31
Inmunoterapia
A partir de que en la hematopoyesis normal existe una interacción entre las células T, las citoquinas, la inmunidad innata y las células estromales; la desregulación del sistema inmune también está implicada en la fisiopatología de los SDM. Además, se ha descrito la asociación con autoinmunidad en pacientes con bajo riesgo, por lo que algunos inmunomodulares, como la lenalidomida, se usan en el tratamiento de este tipo de pacientes.
Diferentes citosinas se expresan de forma anómala en los SMD (interferón gamma, factor de necrosis tumoral alfa y beta, factor β transformador del crecimiento, etc.). Estas de alguna manera inducen la expresión de otras moléculas relacionadas con la muerte celular programada como PD-1 (receptor de la muerte celular programada), PD-L1 (ligando de la muerte programada) y CTLA-4 (antígeno de linfocitos T citotóxico- 4) modulan la activación de las células T y la participan en la vigilancia antitumoral en las neoplasias mieloides. Estas moléculas se han observado sobre expresadas en los SMD, mayormente después de uso de los AHM.32
Por estas razones, se están realizando ensayos con anticuerpos monoclonales dirigidos hacia estos puntos como son el Pembrolizumab (PD-1 inhibidor), el Ipilimumab (CTLA-4 inhibidor) y los inhibidores PD-L1 (Atezolizumab y Durvalumab), medicamentos ya aprobados para otras malignidades que han mostrado efectividad en tumores refractarios a la quimioterapia estándar
En los SMD refractarios se han comunicado resultados limitados como monoterapia y se hacen nuevos ensayos combinándolos con otras drogas, fundamentalmente AHM.32
Inhibidores del espliceosoma
Las mutaciones de genes involucrados en la maquinaria del espliceosoma (SF3B1, SRSF2 y U2AFI) se detectan en un elevado porcentaje (45-85 %) de los SMD y están relacionadas con su patogenia. SF3B1 tiene una elevada incidencia en pacientes con anemia refractaria con sideroblastos anillados con trombocitosis, mientras que SRSF2 y U2AFI son más frecuentes en la leucemia mielomonocítica crónica y las variantes con exceso de blastos y se asocian con un peor pronóstico.11,33 Estas mutaciones son mutuamente excluyentes.
Estudios in vitro revelan la inducción de apoptosis mediada por caspasas, en una línea celular de páncreas humano que recibieron tratamiento con H3B-8800, modulador de SF3B1 de uso oral que actualmente está en ensayo clínico.34
Finalmente, una formulación liposomal de citarabina y daunorubicina (CPX-351) fue aprobada en el año 2018 para el tratamiento de la LMA secundaria y post SMD y pudiera ser una opción en pacientes con SMD de alto riesgo y fallo de los AHM.35
A pesar de la frecuencia creciente de los casos con diagnóstico de SMD y de los múltiples esfuerzos de la comunidad científica, lamentablemente los pacientes que no responden a los AHM tienen pocas opciones de tratamiento, sobre todo los casos de alto riesgo. La opción actualmente más empleada es la inclusión de los pacientes en ensayos clínicos experimentales con alguna de las drogas descritas u otras que puedan dirigirse contra una anormalidad específica; el hándicap de los costos de estos limita el acceso fuera de los ensayos clínicos.
La terapia post AHM, fundamentalmente en los pacientes refractarios o en recaída, tiene muchas alternativas, la mayoría con número reducido de casos y resultados limitados. El futuro va encaminado a la individualización con base en las alteraciones moleculares que puedan ser identificadas en los enfermos con esta entidad, lo que indica que la medicina (diagnóstico y tratamiento) de precisión es la solución en esta y muchas otras malignidades, para poder dirigir el tratamiento a la diana específica.

