










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Las leucemias agudas de linaje ambiguo (LALA) constituyen un grupo heterogéneo de leucemias agudas que no muestran una evidencia clara de diferenciación a lo largo del linaje celular.1 Representan alrededor de 2 a 5 % del total de leucemias agudas. Tienen una incidencia anual de 0,35 casos por un millón de personas. Afectan con mayor frecuencia al sexo masculino y muestran una distribución bimodal de la edad, con picos en menores de 19 años y en mayores de 60. La edad promedio de presentación es alrededor de los 50 años, menor que la leucemia mieloide aguda (LMA) y mayor que la leucemia linfoide aguda (LLA).1
El diagnóstico definitivo de este tipo de leucemia se realiza exclusivamente por las características inmunofenotípicas, con el uso de la citometría de flujo multiparamétrica. De ahí la importancia de actualizar los criterios diagnósticos, la clasificación, el manejo clínico y terapéutico de esta variedad de leucemias agudas.
Métodos
Se realizó una búsqueda de información científica relacionada con el tema en libros de textos de Hematología Clínica y en artículos publicados a través de PUBMED, en los últimos 10 años. Se utilizaron como palabras clave: leucemia aguda; linaje ambiguo; citometría de flujo. Se hizo un análisis y resumen de la bibliografía; se tomaron los aspectos más importantes referidos al tema.
Análisis y síntesis de la información
Al igual que en el resto de las leucemias agudas en esta variedad debe existir en la médula ósea un porcentaje igual o mayor que 20 de blastos. Las características morfológicas de las células leucémicas procedentes de los pacientes con LALA no muestran uniformidad. Pueden encontrarse de forma simultánea blastos de diferentes tamaños y estados de maduración. Estos pueden presentar apariencia linfoide, mieloide, indiferenciada o mixta (Fig.1).2
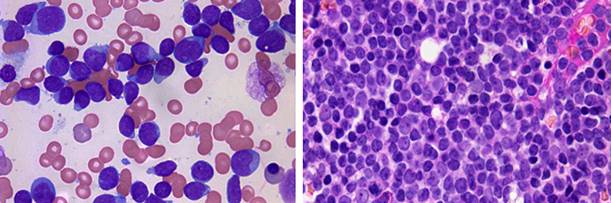 Fuente: Porwit A, Bene MC. Multiparameter Flow Cytometry Applications in the Diagnosis of Mixed Phenotype Acute Leukemia. Cytometry Part B (Clinical Cytometry) 2019;96B:183-94.
Fuente: Porwit A, Bene MC. Multiparameter Flow Cytometry Applications in the Diagnosis of Mixed Phenotype Acute Leukemia. Cytometry Part B (Clinical Cytometry) 2019;96B:183-94.Fig. 1 Citología de la médula ósea. A) Morfología del aspirado de la médula ósea y B) Biopsia de paciente con leucemia aguda de fenotipo mixto linfoide/mieloide.
El diagnóstico definitivo se realiza exclusivamente por las características inmunofenotípicas, con el uso de la citometría de flujo multiparamétrica (CFM).3) Es necesario realizar el análisis de un número igual o mayor que 100 blastos y un total de igual o mayor que 20 000 eventos por cada tubo analizado.
Para la determinación del fenotipo leucémico se debe emplear un panel de anticuerpos monoclonales dirigidos contra los antígenos linfoides B y T, mieloides y precursores hematopoyéticos indiferenciados.
Panel de linaje B: CD10, CD19, CD20, CD22, CD79a citoplasmático, CD79b, HLA-DR, cadena pesada µ citoplasmática e inmunoglobulina de superficie.
Panel de linaje T: CD1, CD2, CD3 citoplasmático y de superficie, CD4, CD5, CD7 y CD8.
Panel mieloide: mieloperoxidasa citoplasmática, CD13, CD14, CD15, CD33, CD41, CD42, CD61, CD64, CD71 y CD235a.
Panel de precursores hematopoyéticos indiferenciados: CD133, CD34, CD38, HLA-DR y CD117.
Debe estar presente en todos los paneles el antígeno panleucocitario CD45, el cual permitirá la separación y el análisis de las diferentes poblaciones celulares.
Clasificación
En 1995, el grupo europeo para la clasificación de las leucemias agudas (EGIL)4 emitió un sistema de puntuación para la caracterización fenotípica de las leucemias híbridas, basado en la expresión de los diferentes antígenos de diferenciación celular, según la especificidad de linaje, la cual fue adoptada en el año 2001 por la Organización Mundial de la Salud (OMS).
En el 2016, la OMS realizó una actualización de estos criterios e incluyó, la presencia de los reordenamientos citogenéticos y moleculares específicos.5
En la actualidad, las LALA se clasifican en dos variedades: leucemias agudas indiferenciadas (LAI) y leucemias agudas de fenotipo mixto (LAFM).
En las LAI, los blastos no expresan antígenos de linaje definido, no obstante, sí lo expresan aquellos de los precursores hematopoyéticos inmaduros, tales como: CD133, CD34, CD38, HLA-DR, CD117 y Tdt, respectivamente. Las LAI constituyen leucemias muy raras.
En esta variedad, debe realizarse el diagnóstico diferencial con la LMA-M0 y el subtipo de precursor temprano T. En la variedad M0 con diferenciación mínima debe estar expresado además de los antígenos inmaduros, CD13 o CD33, lo que indica el linaje mieloide y en el subtipo de precursor temprano T, deben coexistir antígenos inmaduros, mieloides y aquellos correspondientes a los primeros estadios de la ontogenia T CD1a-, CD8- y CD5-/débil +. 5
En las LAFM los blastos expresan antígenos de más de un linaje celular y se dividen a su vez, en: bifenotípicas y bilineales (biclonales) o multilineales. 6,7
En las leucemias agudas bifenotípicas se observa una población neoplásica homogénea y los blastos coexpresan antígenos correspondientes a varias líneas de diferenciación celular. La coexpresión antigénica incluye la combinación de marcadores B/mieloide (Fig. 2), T/mieloide (Fig.3), B/T y trilinaje B/T/mieloide.8,9
 Fuente: Charles N, Boyer D. Mixed-Phenotype Acute. Arch Pathol Lab Med 2017;141:1464.
Fuente: Charles N, Boyer D. Mixed-Phenotype Acute. Arch Pathol Lab Med 2017;141:1464.Fig. 2 Leucemia aguda bifenotípica B/mieloide. A) Morfología de la sangre periférica que muestra blastos grandes, con núcleo irregular y algunas vacuolas citoplasmáticas. B) Inmunofenotipo por citometría de flujo que exhibe una población de blastos (azul) que coexpresan antígenos linfoides B CD19++/CD79a citoplasmático+/CD34+/CD45débil+ y antígenos mieloides, tales como: MPO citoplasmática+ y CD15+.
 Fuentes: Wang P, Peng X, Dean X, Gao L, Zhang X, Feng Y. Diagnostic challenges in T-cell lymphoblastic lymphoma, early T-cell precursor acute lymphoblastic leukemia or mixed phenotype acute leukemia. Medicine 2018;97:4.
Fuentes: Wang P, Peng X, Dean X, Gao L, Zhang X, Feng Y. Diagnostic challenges in T-cell lymphoblastic lymphoma, early T-cell precursor acute lymphoblastic leukemia or mixed phenotype acute leukemia. Medicine 2018;97:4.Fig. 3 Leucemia aguda bifenotípica T/mieloide. Inmunofenotipo por citometría de flujo que muestra una población de blastos (rojo) con expresión de los antígenos: CD117+/CD7+débil+/HLA-DR+/CD3citoplasmático+/MPO citoplasmática+.
En las leucemias agudas bilineales (Fig. 4) o multilineales existen dos o más poblaciones leucémicas, cada una con fenotipos particulares.2,6,7
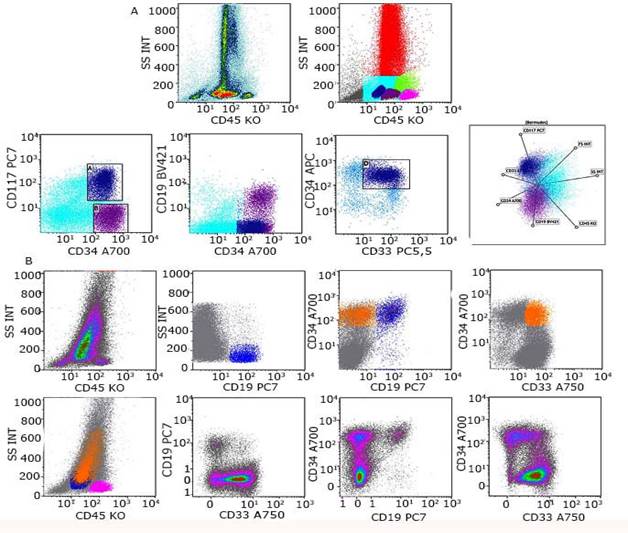 Fuente: Porwit A, Bene MC. Multiparameter Flow Cytometry Applications in the Diagnosis of Mixed Phenotype Acute Leukemia. Cytometry Part B (Clinical Cytometry) 2019;968:183-94.
Fuente: Porwit A, Bene MC. Multiparameter Flow Cytometry Applications in the Diagnosis of Mixed Phenotype Acute Leukemia. Cytometry Part B (Clinical Cytometry) 2019;968:183-94.Fig. 4 Inmunofenotipo de la leucemia aguda bilineal B/mieloide. a) Se muestran dos poblaciones de blastos: mieloide (azul) y linfoide (púrpura). Ambas poblaciones expresan CD34+, pero difieren en la expresión de CD117+ y CD19+, respectivamente. Los blastos mieloides expresan además, CD33+. b) Se muestran dos poblaciones de blastos: linfoide B (azul oscuro) que expresan los antígenos CD34+ y CD19+ y dos poblaciones mieloides (gráfico de contorno) CD33+/CD34+ y CD33+/CD34-, respectivamente.
La suma de estos clones leucémicos debe igualar o superar al 20 % del total de células nucleadas. Estos clones pueden ser linfoides B, T y mieloides, e incluso bifenotípicos. La identificación de determinadas aberraciones fenotípicas es esencial para diferenciar una pequeña población blástica de los precursores normales.6,7,8,9
Una fuerte expresión del antígeno CD19 asociado con al menos otro marcador de linaje B, CD79a o CD22 citoplasmáticos y CD10, CD20 o CD21 de membrana, es suficiente para la atribución de este linaje. Sin embargo, cuando CD19 muestra una débil densidad de expresión deben estar expresados al menos dos de estos antígenos. Constituyen también antígenos específicos de línea linfoide B la cadena pesada µ, las cadenas ligeras к y λ y la inmunoglobulina de superficie que asociada con el antígeno CD79b, conforman el receptor de la célula B. 7,8,9,10
Para la asignación de la estirpe T debe estar expresado el antígeno CD3 en el citoplasma o la superficie celular asociado con al menos otro marcador de este linaje: CD2, CD5, CD4, CD7 u CD8, respectivamente. Las cadenas α/β y Ɣ/ƪ constituyen antígenos específicos de línea T. 7,8,9
Para definir la progenie mieloide es necesario la expresión de la mieloperoxidasa en el citoplasma celular con al menos otro antígeno de estirpe mieloide: CD13, CD14, CD15, CD33, CD41, CD42, CD61, CD64, CD117, CD235a o muramidasa.9
El antígeno inmaduro CD117 no se expresa normalmente en el linaje linfoide, excepto en el precursor temprano T y sí en los precursores mieloides.7,8,9,10
La mayoría de los investigadores reportan una mayor frecuencia de los clones B/mieloides, seguido de los T/mieloides. Por su parte, los clones leucémicos B/T y trilinajes B/T/mieloides son raros.2,6,8,10
Alteraciones citogenéticas y moleculares
La diferenciación normal de los progenitores en un simple linaje celular es el resultado de la regulación de sucesivos eventos de expresión de determinados factores de transcripción, que ocurren en una secuencia específica o a alteraciones epigenéticas añadidas. Los blastos de los pacientes con LALA muestran múltiples alteraciones citogenéticas.11
Las LAI están fundamentalmente relacionadas con alteraciones en los cromosomas 12 y 13 y con el brazo corto del 5.8
En las LAFM con implicación del linaje B, se encuentran con mayor frecuencia las translocaciones t(9;22)/BCR-ABL1 y t((v;11q23)/MLL y cuando involucran al linaje T, la trisomía del cromosoma 4.12
En las leucemias agudas bifenotíficas donde coexisten ambos linajes linfoides B/T, predomina la translocación t(9;17)(p11;q11).8,10
Otras anormalidades citogenéticas encontradas en menor frecuencia, incluyen: dic(7;12), t(X;12), t(1;19) y t(4;11), respectivamente.13
En algunos pacientes se produce un cambio del linaje leucémico tras la quimioterapia, asociada con la translocación KMT2A. Estos casos inicialmente no se presentaron como una LAFM, lo que sugiere que estas lesiones genéticas les confieren el potencial esencial para la plasticidad de linaje.8
Esta translocación aparece con mayor frecuencia en niños especialmente, lactantes. Los blastos de estos enfermos presentan un fenotipo similar al de la LLA de fenotipo B, excepto que un clon pequeño mieloide o monocítico está presente de forma concomitante.10
Algunos autores han encontrado mutaciones en los genes NOTCH cuando están presentes antígenos linfoides T y mieloides. De forma similar, pueden aparecer mutaciones en los genes RAS y TP53, relacionados con comportamientos clínicos más agresivos. Las mutaciones en los genes DNMT3A son más frecuentes en pacientes ancianos.11,12,13
Por su parte, otros investigadores 11) han reportado mutaciones en los genes IKZF1, así como en los reguladores epigenéticos TET2, EZH2 y ASXL1, respectivamente.
Otros estudios8) han revelado mutaciones en genes WT1 (asociados con la célula madre hematopoyética) y en los genes MN (relacionados con la leucemogénesis).
Características clínicas
Los pacientes con LALA presentan manifestaciones clínicas relacionadas con el fallo de la médula ósea, tales como: fatiga, sangramientos e infecciones, producto de la anemia, la trombocitopenia y la disminución tanto en el número como en la función de los leucocitos y por la supresión general que tiene lugar en el sistema inmunológico.14
Se presentan comúnmente con un conteo normal o aumentado de leucocitos en la sangre periférica, hiperviscosidad y alto grado de infiltración leucémica extramedular.15
Pronóstico
Las LALA constituyen un subtipo de leucemias agudas de alto riesgo, con un pronóstico desfavorable y una pobre supervivencia global (SG) de los enfermos.15
Son considerados como factores de riesgos la edad, el conteo de leucocitos en sangre periférica al inicio de la enfermedad, las alteraciones citogenéticas adversas, la creatinina, el ácido úrico, la infiltración extramedular y la respuesta al tratamiento de inducción de la remisión.11,12,15
Tratamiento
Debido a la versatilidad en los criterios diagnósticos y a las pocas series de pacientes publicadas, aún no existe consenso en los protocolos de tratamiento que se emplean en esta variedad de leucemias agudas.16
La mayoría de los grupos de trabajo incluyen inicialmente, los corticoesteroides, seguidos de una quimioterapia similar al de la LLA. En ausencia de respuesta a este tratamiento, adicionan una terapia similar a la de la LMA, con agentes alquilantes.12,13 14
Un menor grupo de investigadores emplean la terapia combinada de leucemia linfoide y mieloide, respectivamente.17
Diferentes investigaciones plantean que la remisión completa se obtuvo mayormente con los protocolos de la LLA, con una SG de 75 a 83 %, mientras que con protocolos de LMA se alcanzaron sobrevidas menores, de 28 a 57 %.16,17
Algunos estudios indican que la terapia combinada resultó ser de igual efectiva cuando se comparó con la terapia de LLA, con una remisión completa de 71 % y 64 %, respectivamente, comparada con 31 % en el caso de tratamiento de LMA. (17
En adultos menores de 40 años se recomienda el uso de los protocolos pediátricos. En los enfermos con t(9;22) positiva, se deben indicar los inhibidores de tirosina quinasas. En pacientes con reordenamiento positivo del gen MLL se emplean inhibidores de enzimas modificadoras de histonas. Aquellos enfermos en quienes no ocurre la remisión de la enfermedad son tributarios de trasplante alogénico de progenitores hematopoyéticos preferentemente, una vez alcanzada la primera remisión.18
Otras opciones de tratamiento para los pacientes quimioresistentes son las terapias de rescate de segunda línea con blinatumomab o con los CAR-T, estos últimos para aquellos pacientes cuyos blastos son CD19 negativos.15,16,17,18
Diferentes razones expuestas por varios investigadores7,18 hacen evidente la pobre evolución clínica de este subgrupo de leucemias agudas, entre las que se encuentran: el fenotipo mixto puede indicar que la célula madre leucémica constituye un progenitor multilinaje primitivo quimiorresistente con lenta replicación, los blastos con fenotipo mixto pueden adaptarse al tratamiento, cambiando el fenotipo celular y que algunas LAFM expresan altos niveles de proteínas de resistencia multidrogas.
La revisión detallada y el reporte oportuno de las LALA son indispensables para aunar los criterios necesarios para perfeccionar el diagnóstico, la clasificación y la toma de decisiones terapéuticas en este grupo de pacientes.

