










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La Organización Mundial de la Salud estima que cerca del 7 % de la población mundial posee alteraciones genéticas en la molécula de hemoglobina (Hb). En países subdesarrollados la mortalidad infantil y neonatal es alta por desnutrición e infecciones, y aún no se reconoce como un importante problema de salud pública. Sin embargo, una vez que la condición económica mejora y la tasa de mortalidad infantil disminuye, las alteraciones genéticas de la molécula de Hb comienzan a ser una carga para los servicios de salud, fenómeno que se observa en varias partes del mundo. Como resultado de las migraciones, estas condiciones se observan con frecuencia en países en los que no se les conoce previamente. Algunas de estas, como la anemia drepanocítica y las formas más graves de talasemia constituyen emergencias médicas, causas de discapacidad, estrés familiar y altos costos económicos.1
Los defectos genéticos de la Hb son un grupo de enfermedades autosómicas recesivas que se dividen en defectos en los que existe una tasa reducida de producción de una o más moléculas de globina: las talasemias; y aquellos en los se producen cambios estructurales que conducen a inestabilidad o anormal transporte de oxígeno. Como todas las clasificaciones, esta no es enteramente satisfactoria, algunas variantes estructurales como por ejemplo la HbE es sintetizada en pocas concentraciones y produce un cuadro típico de talasemia.2
En este artículo se explican los diferentes mecanismos por los cuales ocurren las talasemias y otras alteraciones en la síntesis de las cadenas de globina, así como las características moleculares, fisiopatogénicas y los cambios hematológicos.
Métodos
Se realizó una revisión de la literatura, en inglés y español, en las bases de datos PubMed, ScienceDirect, SciElo y el motor de búsqueda Google académico, de artículos publicados en los últimos 10 años sobre alteraciones en la síntesis de las cadenas de globina. El 78,4 % de los trabajos seleccionados fueron artículos originales y de revisión publicados entre los años 2015-2019. Se hizo un resumen de la bibliografía y se analizaron los aspectos más importantes referidos al tema.
Análisis y síntesis de la información
Talasemias y defectos relacionados
Las talasemias son un grupo heterogéneo de defectos genéticos en la síntesis de Hb, que causan la disminución en la tasa de producción de una o más cadenas de la molécula. Se dividen en α-,β-, δβ- o γδβ-talasemias, de acuerdo a la cadena de globina que presenta el defecto. En algunas, las cadenas de globina no se sintetizan o están ausentes y son llamadas α0- o β0-talasemias; en aquellas que existe una disminución en la concentración de la cadena se nombran α+- o β+-talasemias. Las δβ-talasemias también se subdividen de este modo (Fig. 1).2,3
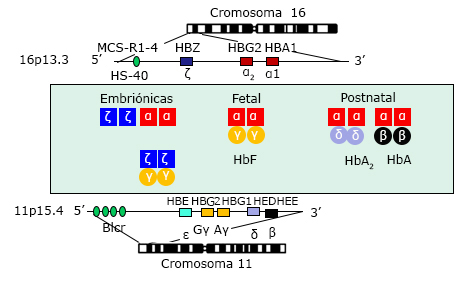 Genes embrionarios y fetales se indican como cajas abiertas.Genes que permanecen activos durante la vida postnatal se señalan en gris y negro. Diferentes hemoglobinas expresadas durante el período embrionario, de izquierda a derecha Gower-1 (ζ2 ε2), Gower-2 ( α2ε2) y Portland (ζ2 γ2), período fetal (HbF) y período postnatal (HbA y HbA2).Fuente: Farashi S, Harteveld CL. Molecular basis of α-thalassemia. Blood Cells Mol Dis. 2018; 70:43-53)
Genes embrionarios y fetales se indican como cajas abiertas.Genes que permanecen activos durante la vida postnatal se señalan en gris y negro. Diferentes hemoglobinas expresadas durante el período embrionario, de izquierda a derecha Gower-1 (ζ2 ε2), Gower-2 ( α2ε2) y Portland (ζ2 γ2), período fetal (HbF) y período postnatal (HbA y HbA2).Fuente: Farashi S, Harteveld CL. Molecular basis of α-thalassemia. Blood Cells Mol Dis. 2018; 70:43-53)Fig. 1 Ubicación cromosómica de los grupos de genes α - y β-globínicos en los cromosomas 16p y 11p.
Las talasemias ocurren en aquellas poblaciones que presentan variaciones estructurales en la molécula de Hb, por lo que es común encontrar individuos que hereden un gen talasémico de un padre y un gen con alteraciones estructurales del otro progenitor. Estas interacciones producen defectos genéticos, clínicamente diversos con un rango de gravedad desde anemias hipocrómicas ligeras y asintomáticas hasta muerte in útero.3
(-talasemias
Las (-talasemias se extienden por toda el África subsahariana, la región del Mediterráneo, Medio Oriente, el subcontinente indio, desde el sur de China hasta Tailandia, el archipiélago malayo, Indonesia hasta las islas del pacífico.4
Herencia y clasificación
La HbA y la HbF presentan cadenas ( y defectos en su síntesis provoca defectos en la producción de estas Hb. La deficiencia de cadenas ( en el feto lleva a la producción en exceso de cadenas ( y de esta forma tetrámeros-(4 o HbBart; en el adulto, se produce un exceso en cadenas ( que da lugar a tetrámeros-(4 o HbH (Fig.2).3,4
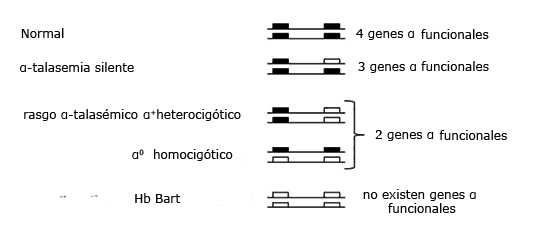 Fuente: Farashi S, Harteveld CL. Molecular basis of α-thalassemia. Blood Cells Mol Dis. 2018; 70:43-53.
Fuente: Farashi S, Harteveld CL. Molecular basis of α-thalassemia. Blood Cells Mol Dis. 2018; 70:43-53.Fig. 2 Expresión fenotípica y clasificación de los defectos α talasémicos.
Existen dos genes (-globina en el cromosoma 16, por lo tanto dos tipos principales de (-talasemia: α0-talasemia, en la que ambos genes están inactivados (-/αα) y α+-talasemia, en la que uno está afectado (-α/αα). De aquí que existan dos entidades clínicas bien definidas: el síndrome del hidrops fetalis por Hb Bart y la enfermedad por HbH.5,6
Biología molecular
La α0-talasemia se produce por deleciones en ambos genes (-globínicos y también puede ser resultado de la deleción de 40 kb "corriente arriba" del gen (-globínico que involucra la región HS40. La α+-talasemia es un poco más compleja y se produce por la remoción de uno del par de genes (-globínicos, y deja el otro par intacto (-α/αα). En otros, ambos genes están intactos pero uno de ellos presenta mutaciones que inactivan parcial o totalmente la actividad de estos (αTα/αα).7
Cada gen ( yace dentro de un límite de 4 kb aproximadamente, generadas por duplicación y subsecuente división, y produce tres subsegmentos homólogos nombrados X, Y y Z. Las cajas Z duplicadas son de 3,7 kb y las cajas X de 4.2 kb. La desalineación de estos segmentos y el entrecruzamiento recíproco durante la meiosis produce cromosomas con un solo gen (-α) y otro con el gen triplicado (ααα). Si el entrecruzamiento ocurre entre cajas Z, 3.7 kb del ADN se pierden y se denomina deleción derecha (-α3.7); si ocurre entre dos cajas X se pierden 4.2 kb y se denomina deleción izquierda (-α4.2).8
En las formas sin deleción los genes están intactos, pero uno de ellos está inactivado por mutaciones; es común la sustitución de una base nitrogenada en el codón de parada UAA por CAA que codifica para glutamina. Cuando los ribosomas alcanzan este punto no terminan la cadena y el ARNm es "leído" hasta que se alcance otro codón de parada. Una cadena muy elongada es producida a una tasa muy reducida. Esta variante se nombra Hb "Constant Spring", donde se reúnen otras variaciones provocadas por el cambio del codón de terminación.9
Relación genotipo-fenotipo
El síndrome del hidrops fetalis por Hb Bart se produce por la herencia homocigótica de α0-talasemia. Otras formas de la enfermedad por HbH se produce por la herencia de α0- y α+-talasemia y la herencia conjunta de α0-talasemia y de la Hb “Constant Spring”.10
La homocigocidad de las formas sin deleción de la α+-talasemia presenta fenotipos más graves que las formas con deleción y puede resultar en la enfermedad por HbH. La homocigosis para las formas con deleción (-α/-α) se presentan con una anemia hipocrómica ligera muy similar a los estados heterocigóticos para la α0-talasemia.11
Fisiopatología
La deficiencia de cadenas ( conduce a la producción en exceso de cadenas ( y (, que forman la Hb Bart y la HbH, respectivamente. Los tetrámeros formados no precipitan en la médula ósea por lo que la eritropoyesis es más efectiva. Sin embargo, la HbH es inestable y precipita en los eritrocitos a medida que estos envejecen, forma cuerpos de inclusión junto a daños a la membrana provocados por el hemo y el hierro. Estos eritrocitos son atrapados en el bazo y en la microcirculación, con la consiguiente disminución de su sobrevida. Estas Hb presentan alta afinidad por el oxígeno cuya curva de disociación se asemeja al de la mioglobina.4
Síndrome del hidrops fetalis por HbBart
Es causa de muerte fetal. No existe producción de cadenas ( y por lo tanto, ni de HbF, ni de HbA. Los fetos nacen entre las 28-40 semanas, y si nacen vivos jadean por unos minutos y mueren dentro de la primera hora de nacidos. Los neonatos muestran cuadro típico de hidrops fetalis con palidez extrema, anasarca y hepatoesplenomegalia masiva, anormalidades congénitas y placenta grande desmenuzada. Presentan cambios talasémicos graves en los eritrocitos, Hb entre 60-80 g/L y numerosas células nucleadas. La Hb presenta 80 % de Hb Bart y 20 % de Hb Portland (ζ2γ2). Estos fetos llegan a término por la producción continuada de las Hb embrionarias. El síndrome se caracteriza por una alta incidencia de toxemia gravídica y complicaciones obstétricas provocadas por el gran tamaño de la placenta.12
Enfermedad por HbH
Se caracteriza por la presencia de anemia y esplenomegalia. Los rasgos talasémicos graves y retardo en el crecimiento son infrecuentes. Los pacientes alcanzan la vida adulta con episodios de hemólisis grave exacerbados por infecciones y progresivo hiperesplenismo. La Hb se mantiene entre 70-100 g/L y los extendidos periféricos muestran características talasémicas, reticulocitosis moderada, numerosos cuerpos de inclusión por la precipitación de la HbH bajo la acción redox de colorantes supravitales. Los análisis de Hb revelan 5-40 % HbH, HbA y HbA2 normal o reducida.13
Rasgos (-talasémicos
Los rasgos α0-talasémicos son anemia hipocrómica ligera y HbA2 normal. No existen ensayos de rutina para el diagnóstico de esta condición, excepto los ensayos moleculares. Los portadores de α+-talasemia delecional presentan cuadro hematológico similar al normal. Los estados heterocigóticos de las formas sin delaciones a veces están asociados con anemia ligera; el tipo asociado con la Hb “Constant Spring” se identifica por la presencia de trazas de variantes de Hb en electroforesis alcalina.14
Los portadores α0-talasemia son identificados con precisión en el período neonatal cuando presentan entre 5-10 % de Hb Bart que luego desaparece y no es reemplazada por la HbH. Los portadores de α+-talasemia tienen ligero incremento de Hb Bart (1-3 %) pero su ausencia no excluye el diagnóstico.15
Otras formas de (-talasemia
Otras α-talasemias no están relacionadas en su patogénesis y distribución con las formas descritas anteriormente. Estas incluyen el síndrome α-talasémico de retardo mental y la asociación de la α-talasemia con la mielodisplasia.
El síndrome α-talasémico de retardo mental se presenta en dos formas: uno codificado en el cromosoma 16 (ATR-16) y el otro en el cromosoma X (ATRX). En el ATR-16 hay pérdida de dos megabases del extremo subtelomérico del cromosoma 16p. Los niños presentan retardo mental ligero sin características dismórficas.16) El ATRX está relacionado con características dismórficas y retardo mental grave. Las mutaciones en este cromosoma producen alteraciones en las ADN- helicasas, en factores de transcripción involucrados en el modelaje de la cromatina y la regulación de genes. El producto de estos genes tiene una función importante en la transcripción de los genes α-globínicos y muchos otros genes durante el desarrollo temprano.17
El fenotipo α-talasémico se asocia también con mielodisplasia en pacientes ancianos y esta condición es igualmente causada por mutaciones en el gen ATRX. En sangre periférica se observan características dismórficas con poblaciones de eritrocitos con niveles variables de HbH y cuerpos de inclusión.18
(-talasemias
Las β-talasemias son los tipos más importantes de talasemia por ser frecuentes y producir anemia grave en los estados homocigóticos y heterocigóticos. Se distribuyen por la zona del Mediterráneo y África del norte y oeste, Medio Oriente, la India y el sureste asiático. Las zonas de alta incidencia se ubican en Yugoslavia, Rumania, región sur de la antigua Unión Soviética, China, Tailandia, Malacia, Indonesia y algunas islas del pacífico; la una frecuencia génica es entre el 2-30 %. La β-talasemia no está confinada estrictamente a estas regiones y ocurre en todos los grupos poblacionales.1,19
Biología molecular
Las mutaciones que causan β-talasemia involucran todas las etapas de producción de la cadena globínica: transcripción, traducción y en la estabilidad postraduccional del producto génico (Fig. 3).20
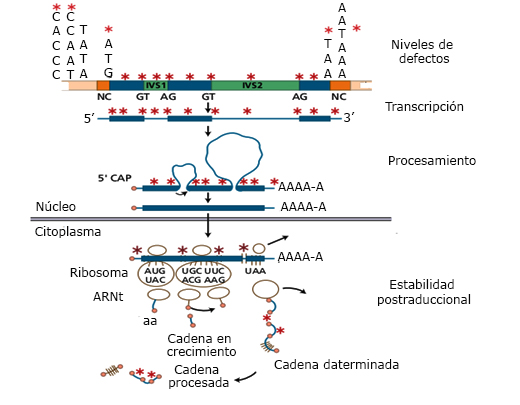
Transcripción
La transcripción es afectada por deleciones y puntos de mutaciones en la región promotora del gen β-globínico con la excepción de la deleción de aproximadamente 600 bases del extremo 3´ del gen, restricto para ciertas poblaciones de la India, deleciones mayores son infrecuentes. Las mutaciones en la región promotora y en las regiones adyacentes subregulan el gen en grado variable causando diversas formas de β-talasemias.21
Procesamiento
Las mutaciones dentro de los exones e intrones o en sus uniones interfieren con el mecanismo de empalme de los exones luego de que el intrón es removido. La sustitución de bases en las secuencias invariantes GT o CT en la unión exón-intrón impide el empalme y es causa de β0-talasemia. La secuencia adyacente a GT en los intrones es relativamente conservada y se denomina secuencia consenso. Las mutaciones en esta región u otras de los intrones se asocian con defectos en la producción de la cadena β-globínica; se producen sitios de empalmes alternativos y la consiguiente síntesis de especies normales o anormales de ARNm. El ARNm incorrectamente empalmado no es funcional porque contiene secuencias de intrón que generan mutaciones sin sentido o cambios en el marco de lectura. En los exones también existen secuencias consenso en la unión intrón-exón y mutaciones en estas secuencias también conducen a un empalme anormal.22
Traducción
Estas mutaciones se dividen en dos grupos: mutaciones sin sentido que producen codones de parada en la porción codificadora del ARNm y terminación prematura de la síntesis de la cadena, pero como parte de los mecanismos de control del procesamiento del ARNm, el mensaje no es transferido al citoplasma celular, fenómeno denominado “alineamiento sin sentido”. El segundo, mutaciones en los exones que producen alteraciones en el marco de lectura por pérdida o inserción de una o más bases.23
Estabilidad postraducción
Las mutaciones sin sentido en el exón 3 no están sujetas a “alineamiento sin sentido”, por lo que los ARNm anormales son transportados al citoplasma y traducidos, lo que puede resultar en productos inestables que forman inclusiones en los precursores eritroides. En otros casos, las cadenas β muy inestables forman moléculas de Hb viables, pero los eritrocitos son rápidamente eliminados de la circulación conduciendo a una anemia hemolítica crónica.24
Fisiopatología
Los defectos moleculares en la β-talasemia resultan en ausencia o reducción de la producción de la cadena β-globínica. La síntesis de las cadenas α no es afectada y se produce un desbalance que provoca un exceso de las cadenas α. En ausencia de su cadena “hermana”, esta precipita y da lugar a inclusiones intracelulares que interfiere con la maduración de los eritrocitos. Ello provoca destrucción intramedular de estos precursores que conduce a una eritropoyesis ineficaz.25
Los eritrocitos que maduran y entran en la circulación contienen inclusiones de cadenas α que interfieren con su paso por la microcirculación, especialmente en el bazo. Los productos de degradación de las cadenas α en exceso, particularmente hierro y hemo, producen efectos deletéreos en las proteínas y lípidos de la membrana eritrocitaria, que se manifiesta en anomalías en la homeóstasis de electrolitos y en la deformabilidad de la membrana. El resultado final es la rigidez de la membrana del eritrocito y el acortamiento de su sobrevida.26
La anemia es el resultado de una eritropoyesis ineficaz y de la hemólisis. Se estimula la producción de eritropoyetina que causa expansión medular y serias deformidades de los huesos largos. El bazo se hipertrofia provocando esplenomegalia, que junto a la expansión medular causa un incremento en el volumen plasmático, contribuyendo a la anemia.27 La producción de HbF disminuye después del nacimiento, pero algunos precursores eritroides adultos (células F) retienen la habilidad para producir cadenas ( que se combinan con el exceso de cadenas α para formar la HbF.20
Las células con mayor número de cadenas ( son protegidas de la precipitación de las cadenas α y seleccionadas en la médula ósea y en sangre periférica por lo que la HbF es encontrada en los eritrocitos. La síntesis de las cadenas ( no se afecta, el desorden se caracteriza por un incremento en la producción de HbA2 (α2(2) (Fig. 4).21
Si la anemia es corregida con transfusiones de eritrocitos, la producción de eritropoyetina es interrumpida, el crecimiento y desarrollo son normales, las deformidades óseas no ocurren y la esplenomegalia es menos marcada. Si se tiene en cuenta que cada unidad de sangre transfundida contiene aproximadamente 200 mg de hierro; en los regímenes de hipertransfusión el hierro se acumula en hígado, glándulas endocrinas y miocardio. Aunque los niños talasémicos tienen un desarrollo y crecimiento normales, estos mueren por la hemosiderosis, a menos que esta sea removida (Fig.4).20,27

Relación genotipo-fenotipo
Los modificadores genéticos de los fenotipos β-talasémicos pueden ser divididos en primarios, secundarios y terciarios. Los primarios son mutaciones que provocan efectos variables en la expresión del gen de la β-globina que pueden afectar el rendimiento útil de las cadenas β-globínicas en un rango de cero a muy ligera reducción. Los secundarios reducen el grado de desbalance en la síntesis de las cadenas globínicas e incluyen la herencia de α-talasemia y una variedad de modificadores de la producción de las cadenas ( en la vida adulta.28) Los terciarios son responsables de las complicaciones de la enfermedad: la gravedad de la enfermedad ósea, la hemosiderosis, la ictericia y la propensión a infecciones por el polimorfismo que involucra el sistema inmune y su regulación.29,30
Hallazgos clínicos
Las formas graves se presentan en el primer año de vida con dificultades en el crecimiento, inapetencia, infecciones y toma del estado general; los infantes se vuelven pálidos y la esplenomegalia está bien establecida. No existen signos específicos y el diagnóstico depende de los cambios hematológicos. Si el niño recibe transfusiones de forma regular, el desarrollo temprano es normal hasta la pubertad, cuando los efectos de la hemosiderosis empiezan a aparecer. Si el niño no es transfundido adecuadamente, empiezan aparecer las características típicas del cuadro clínico de la β-talasemia.31
Sin una adecuada terapia quelante de hierro, los efectos de la hemosiderosis comienzan al final de la primera década de vida.32) El crecimiento se afecta y aparecen afecciones hepáticas, endocrinas y cardíacas que incluyen diabetes, hipoparatoidismo e insuficiencia adrenal; además de retraso en el desarrollo sexual que puede conducir a serios problemas psicológicos.33) Estos pacientes mueren de forma súbita por arritmias agudas intensificadas por infecciones.34
Los niños con inadecuada terapia transfusional presentan complicaciones en la niñez temprana, además del retardo en el crecimiento y desarrollo. La esplenomegalia progresiva causa anemia que puede asociarse con trombocitopenia y tendencia hemorrágica.35) Producto de la expansión medular se producen deformidades esqueléticas con marcadas protuberancias y sobrecrecimiento del cigoma, causa de las “facies mongoloides” en la β- talasemia; con cambios radiológicos en huesos largos y falanges y la típica apariencia de “pelos de punta”. Estos cambios se acompañan con fracturas recurrentes.36,37
Como resultado de la anemia crónica y la expansión medular, estos niños se convierten en hipermetabólicos con incrementados requerimientos de ácido fólico y depleción aguda de folatos que empeora la anemia. La hiperuricemia y la gota secundaria al recambio de los precursores eritroides ocurre ocasionalmente.38) La trombocitopenia como producto del hiperesplenismo puede ser exacerbada por el daño hepático.35 También pueden desarrollar complicaciones dentarias como dientes pobremente formados, maloclusión e inadecuado drenaje sinusal que conduce a infecciones crónicas y sordera. Si el niño sobrevive la pubertad, desarrolla las mismas complicaciones de la hemosiderosis que desarrollan los niños transfundidos adecuadamente, como resultado de la tasa de absorción gastrointestinal aumentada.39
Cambios hematológicos
Los valores de Hb oscilan entre 20-80 g/L. Se observan eritrocitos con marcada hipocromía, poiquilocitos, anisocromía moderada, muchos macrocitos hipocrómicos, fragmentocitos y punteado basófilo; además, normoblastos y reticulocitos aumentados después de practicada la esplenectomía. Los leucocitos y plaquetas se encuentran normales, a menos que exista hiperesplenismo. La médula ósea presenta una tasa mieloide/eritroide ≤ 1. En muchos precursores eritroides se observan inclusiones luego de incubación con metil-violeta; similares inclusiones son observadas en periferia posterior a la esplectomía. Además, se observa: bilirrubina elevada, haptoglobina ausente, hierro sérico aumentado, capacidad de unión de hierro elevada (en niños dependientes de transfusiones), ferritina auentada. En la biopsia hepática se observan altos niveles de hierro en células parenquimatosas y reticuloendoteliales.26
Cambios hemoglobínicos
Los niveles de HbF se encuentran elevados y heterogéneamente distribuidos entre los eritrocitos. En la β0-talasemia no hay presencia de HbA, mientras que en la β+-talasemia se encuentra entre el 30-90 %. La HbA2 no tiene valor diagnóstico porque generalmente se encuentra en un rango normal como reflejo de la heterogenicidad de las poblaciones celulares en periferia.40
β-talasemia heterocigótica
Los portadores de β-talasemia no presentan síntomas, excepto en períodos de estrés como el embarazo, cuando madres se vuelven más anémicas. La esplenomegalia es rara y los valores de Hb se encuentran entre 90-110 g/L. En periferia se observa ligera hipocromía y microcitosis, reliculocitos en un rango normal. En médula hay moderada hiperplasia eritroide. Un hallazgo característico es la elevación de la HbA2 (4-6 %) y una ligera elevación de la HbF (1-3 %), en el 50 % de los casos.41
β-talasemia asociada a variantes hemoglobínicas
En poblaciones que presentan alta incidencia de β-talasemia y varias variantes hemoglobínicas, es común la herencia del gen β-talasémico de un progenitor y una variante hemoglobínica del otro. Aunque muchas de las interacciones han sido descritas, las más comunes son: HbSβ-talasemia, HbCβ-talasemia y HbEβ-talasemia. Existen más de 300 variantes hemoglobínicas, las más frecuentes se presentan en el anexo.
En poblaciones africanas se observan formas ligeras de β-talasemia cuando interaccionan con el gen de la HbS. Se caracteriza por anemia ligera, pocas crisis drepanocíticas y sobrevida normal. En poblaciones mediterráneas el fenotipo HbSβ0 o β+-talasemia es indistinguible de la anemia drepanocítica. En pacientes HbSβ+-talasemia el diagnóstico se confirma por la electroforesis de Hb, además de la HbS se observa 5-30 % de HbA y valores elevados de HbA2 y los pacientes HbSβ0-talasemia, altos niveles de HbF y HbA2.42
HbCβ-talasemia
Se caracteriza por anemia ligera y esplenomegalia. En periferia se observan eritrocitos en diana y reticulocitos moderadamente elevados. La electroforesis de Hb muestra predominio de la HbC. El diagnóstico se confirma detectando el rasgo HbC en un progenitor y el talasémico en el otro.43
HbEβ-talasemia
La HbE es sintetizada ineficientemente y por lo tanto cuando se hereda junto a la β0-talasemia existe marcada deficiencia en la producción de las cadenas β. Los cambios clínicos y hematológicos son variables con anemia grave, esplenomegalia, cambios talasémicos en huesos y valores de Hb entre 4-9 g/L. En médula ósea se observa marcada hiperplasia eritroide con inclusiones de cadenas α en muchos precursores. Las complicaciones incluyen: infecciones, hiperesplenismo secundario, hemosiderosis, lesiones neurológicas, deficiencia de folato y fracturas recurrentes.44
Algunos pacientes presentan crecimiento y desarrollo normal con pocas complicaciones. El diagnóstico se confirma con el hallazgo de HbE y HbF en la electroforesis de Hb. En casos de HbE β+-talasemia, pequeñas cantidades de HbA están presentes.45
Formas de β-talasemia
Un subgrupo de β-talasemia heterocigótica presenta valores de HbA2 > 8 % del total de la Hb y es el resultado de pequeñas deleciones en el extremo 5´terminal del gen de la cadena β que incluye la región promotora. El cuadro clínico y hematológico es idéntico a las formas comunes de β-talasemia. Algunos individuos tienen niveles normales de HbA2 por la herencia del rasgo (-talasémico. Es una forma ligera de β-talasemia que es completamente silente en heterocigotos. En ocasiones sigue un patrón de herencia dominante y los heterocigotos se vuelven sintomáticos. El cuadro clínico se caracteriza por: anemia moderada, esplenomegalia con marcados rasgos talasémicos, eritropoyesis ineficaz y cuerpos de inclusión intracelulares.46
Los pacientes no son dependientes de transfusiones y el daño hepático y endocrino producto de la hemosiderosis puede estar disminuido. Algunos presentan mutaciones en el exón 3 del gen β-globínico.46
(β-talasemias
Se producen por deleciones en los genes β y (-globínicos mientras que en otros es producto de sinapsis desapareada y entrecruzamiento desigual en el locus de los genes β y (-globínicos con la producción de genes (β fusionados y por consiguiente cadenas (β fusionadas que combinadas con las cadenas α forma una variante de Hb denominada Hb“Lepore” (es el nombre de la familia donde se identificó por primera vez). Esta condición puede ser clasificada en ((β)0 talasemia y Hb“Lepore” ((β)+- talasemia.47
La ((β)0 talasemia resulta de deleciones de diferentes longitudes en el gen β-globínico. En algunos casos los genes G( y A( permanecen intactos, en otros el gen A( se encuentra involucrado por lo que se denominan ((β)0 o (A((β)0 talasemia. Las deleciones dejan los genes (-globínicos activos en la vida adulta y su producción es alta aunque no tanto en fetos, haciendo esta condición relativamente ligera. La Hb“Lepore” es también heterogénea dependiendo del sitio preciso del entrecruzamiento anormal y por lo tanto las variantes estructurales de las cadenas (β fusionadas. La producción de las cadenas ( es baja por lo que clínicamente es más grave.48
Cambios clínicos y hematológicos
Los pacientes homocigóticos presentan anemia ligera, esplenomegalia, Hb entre 80-100 g/L y 100 % de HbF. Los heterocigóticos tienen un cuadro hemático talasémico, HbF entre 5-20 % y niveles normales de HbA2. En los homocigóticos para la Hb“Lepore”, generalmente, el cuadro clínico es similar al β-talasémico. La Hb está constituida por HbF y “Lepore” únicamente.47
Hemoglobina fetal persistente hereditaria (HFPH)
La HFPH se caracteriza por la producción persistente de HbF en la vida adulta sin mayores anormalidades hematológicas. Se produce por deleciones en el locus del gen β-globínico, similar al que causa (β-talasemia. Los homocigóticos presentan 100 % de HbF, cuadro hemático ligeramente talasémico y ligera policitemia como reflejo de la alta afinidad de la HbF por el oxígeno. Los heterocigotos no presentan anomalías con 20-30 % de HbF.49
Otra variante es el resultado de mutaciones en la región promotora “corriente arriba” para los genes globínicos G( y A( que hacen los genes (-globínicos unidos a los genes β-globínicos activos en la vida adulta y son llamados G(β+ y A(β+HFPH. Los heterocigotos tienen niveles altos de HbF (10-15 %), tanto de G( o A( HbF, según la mutación particular involucrada.48
Un tercer tipo se presenta con bajos niveles de HbF (1-5 %). Ocasionalmente es llamado HFPH“Suiza”, por el sitio donde fue reconocido por primera vez. Es una condición heterogénea y en algunos casos no está relacionada con el grupo de genes globínicos. Su importancia radica en que cuando se hereda con β-talasemia o HbS causa la producción de altos niveles de HbF y reduce la gravedad clínica.50
((((-talasemias
Esta condición es el resultado de deleciones en el grupo de genes (-globínicos. No existe síntesis de proteínas por lo que el estado homocigótico no es compatible con la vida. Los recién nacidos heterocigóticos presentan enfermedad hemolítica muy grave con anemia e hiperbilirrubinemia. Si sobrevive el período neonatal, se desarrolla y crece de forma normal. En la vida adulta se presenta un cuadro hematológico del tipo (-talasémico heterocigótico con anemia ligera, microcitos hipocrómicos y HbA2 normal.51
En sentido general, las talasemias y las hemoglobinopatías son las enfermedades hemolíticas hereditarias más comunes en muchas partes del mundo, caracterizadas por complejas interacciones entre anemia, eritropoyesis ineficaz y alteraciones del metabolismo del hierro. El amplio espectro de estas enfermedades puede ser explicado por la distribución geográfica, guerras, migraciones masivas y la consanguinidad. La cronicidad de la enfermedad y los altos costos de tratamiento hacen crucial las estrategias y programas de prevención que gracias a los que la incidencia ha disminuido sustancialmente en muchos países.


