










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El término hemólisis hace referencia a la destrucción de los eritrocitos (ER) y ocurre en un vasto rango de condiciones clínicas fisiológicas y patológicas. Es empleado para definir situaciones donde la vida media de los ER está disminuida por causas mecánicas, tóxicas, autoinmunes e infecciosas. Una tasa de destrucción alta es suficiente para inducir una disminución de hemoglobina (Hb) a valores inferiores a los de referencia, lo que causa la anemia hemolítica (AH). Esta destrucción o pérdida que conduce a la anemia, junto con las formas de hemorragia y secuestro, difieren de las formas de anemias provocadas por daño o noxas en la eritropoyesis como: anemia aplásica, mielodisplasia, mieloptisis, enfermedades malignas hematológicas y las deficiencias de hierro y vitaminas. Las formas de presentación, las alteraciones en los valores de laboratorio y las estrategias de tratamiento, son también diferentes.1
Este trabajo pretende describir los principales marcadores de hemólisis que se encuentran variablemente alterados en las diferentes formas de AH, lo cual permite realizar el diagnóstico diferencial y la evaluación de la eficacia de los diferentes tratamientos.
Métodos
Se realizó una revisión de la literatura, en inglés y español, a través de los sitios web PubMed, Science Direct, Scielo y el motor de búsqueda Google académico de artículos publicados en los últimos 10 años sobre marcadores de AH. Las palabras clave empleadas fueron: anemias hemolíticas, marcadores hemolíticos, hemoglobina, reticulocitos, esquistocitos, deshidrogenasa láctica, haptoglobina, bilirrubina, ferritina, hemosiderinuria. El 69,4 % de los trabajos seleccionados fueron artículos originales y de revisión publicados entre los años 2015-2019. Se hizo un análisis y resumen de la bibliografía, seleccionando los aspectos más importantes referidos al tema.
Análisis y síntesis de la información
El primer paso para distinguir las AH es la diferenciación entre anemias hemolíticas hereditarias o congénitas (AHH) y las anemias hemolíticas adquiridas (AHA), donde una rigurosa anamnesis del paciente orienta adecuadamente en el diagnóstico. A su vez, es esencial diferenciar las formas de presentación aguda y crónica; la hemólisis extravascular de la intravascular y la presencia de signos extrahematológicos. La observación microscópica de los extendidos de sangre periférica, es determinante en el diagnóstico, particularmente las formas congénitas.
Por otra parte, existen formas de anemia que son causadas por varios factores que concomitan al mismo tiempo, por lo que su diagnóstico es un desafío, como puede ser el déficit de vitaminas o dismielopoyesis, alteraciones en lámina periférica y comorbilidades que afectan la forma clínica de presentación, como la enfermedad hepática (disminución de la producción de marcadores hemolíticos) y la enfermedad renal (insuficiente producción de eritropoyetina).
Respecto a las AHA, la prueba de antiglobulina directa (PAD) es la piedra angular para el diagnóstico, al facilitar la distinción de las anemias hemolíticas autoinmunes (AHAI) en: anemias hemolíticas autoinmunes por anticuerpos calientes (~70 % de los casos con PAD positiva por IgG e IgG+C3dg); síndrome de aglutininas frías (SAF, ~20 % de los pacientes con PAD positiva por C3dg) y tipo mixto (< 10 % con PAD positiva por IgG y C3dg y coexistencia de anticuerpos calientes y títulos altos de crioaglutininas).
Sin embargo, existen casos atípicos difíciles de clasificar con PAD negativa y anemias graves refractarias a varias líneas de tratamiento.2 Existen casos muy raros causados por autoanticuerpos IgM con una amplitud térmica muy próxima a la fisiológica (IgM caliente) caracterizado por una anemia muy grave y tasa de mortalidad superior al 20 %.3 Estas anemias también pueden ser clasificadas de acuerdo a su etiología en idiopáticas o primarias y secundarias a síndromes linfoproliferativos, infecciones, inmunodeficiencias, enfermedades autoinmunes, tumores y trasplante de progenitores hematopoyéticos (Fig.)4,5
 Fuente: Tomado y modificado de: Barcellini W, Fattizzo B. Clinical Applications of Hemolytic Markers in the Differential Diagnosis and Management of Hemolytic Anemia. Dis Markers. 2015; 2015:635670)(4
Fuente: Tomado y modificado de: Barcellini W, Fattizzo B. Clinical Applications of Hemolytic Markers in the Differential Diagnosis and Management of Hemolytic Anemia. Dis Markers. 2015; 2015:635670)(4
Fig. - Diagrama de flujo para el diagnóstico diferencial de las anemias hemolíticas. AH: anemia hemolítica; ADC: anemia diseritropoyética congénita; PAD: prueba de antiglobulina directa; AHAI: anemia hemolítica autoinmune; RHT: reacción hemolítica transfusional; HPN: hemoglobinuria paroxística nocturna
Marcadores hemolíticos
Los marcadores de hemolisis son variados y con el desarrollo tecnológico de las últimas décadas su espectro se ha ampliado a reactantes de fase aguda y citocinas; no obstante, estas tecnologías no están al alcance de todos. La mayoría de los laboratorios continúan detectando de rutina marcadores de hemólisis clásicos como: Hb, reticulocitos, esquistocitos, deshidrogenasa láctica, haptoglobina, bilirrubina, ferritina y hemosiderinuria, los cuales no han perdido su vigencia ni importancia diagnóstica en diferentes entidades clínicas4 (Tabla).
Tabla- Marcadores de hemólisis en diferentes enfermedades hemolíticas
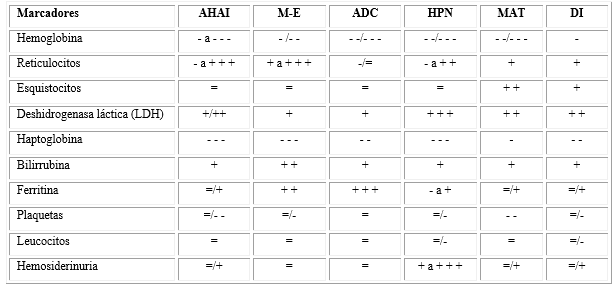
Nota: Los valores están expresados en un estilo semicuantitativo para indicar diferentes intensidades de la alteración en varios síndromes hemolíticos: +/++/+++ indica incremento de ligero a grave; -/- -/- - - indica disminución; = indica valores dentro del rango normal; AHAI: anemia hemolítica autoinmune; M-E: defectos en la membrana y en las enzimas; ADC: anemia diseritropoyética congénita; HPN: hemoglobinuria paroxística nocturna; MAT: microangiopatía trombótica; DI: dispositivo intravascular.
Fuente: Tomado y modificado de: Barcellini W, Fattizzo B. Clinical Applications of Hemolytic Markers in the Differential Diagnosis and Management of Hemolytic Anemia. Dis Markers. 2015; 2015:635670.(4
Hemoglobina
La hemoglobina es el marcador más directo de la gravedad clínica en las enfermedades hemolíticas. Sus valores pueden estar muy próximos a los valores de referencia en las formas ligeras (Hb > 100 g/L) o marcadamente reducidos en las formas moderadas (Hb entre 80-100 g/L), graves (Hb entre 60-80 g/L) y muy graves (Hb < 60 g/L).
En diferentes estudios retrospectivos de AHAI primaria se ha comprobado que el valor de Hb al momento del diagnóstico es el predictor más importante para establecer el criterio de muerte y la necesidad de examinar diferentes líneas de tratamiento.6
La rápida disminución de los valores de Hb, propicia que se produzcan signos relevantes como: taquicardia, astenia y disnea, mientras que la reducción crónica y progresiva es bien tolerada. Generalmente, la destrucción de los ER es mucho más alta en la hemólisis intravascular (200 mL de ER en 1 hora) mientras que en la hemólisis extravascular es 10 veces menor.6
El diagnóstico diferencial involucra de forma frecuente enzimopatías como el déficit de glucosa-6-fosfato-deshidrogenasa (G6PD), diferentes formas de AHAI que involucran la activación del complemento (AHAI caliente [IgG+C3dg], AHAI caliente inducida por anticuerpos IgM, AHAI de tipo mixto), la anemia diseritropoyética congénita (ADC) con un rango térmico muy próximo al fisiológico y la hemoglobinuria paroxística nocturna (HPN).
El curso crónico con crisis agudas es común encontrarlo en membranopatías como la esferocitosis y estomacitosis hereditaria, enzimopatías como la deficiencia de la piruvato-kinasa, AHAI por anticuerpos fríos y en el implante de válvulas cardíacas artificiales o dispositivos intravasculares.6
El monitoreo de los valores de Hb en pacientes hemolíticos debe ser riguroso y deberá tenerse en cuenta para evaluar la respuesta al tratamiento. En las AHAI la respuesta al tratamiento se define como completa para valores de Hb > 120 g/L y normalización en los marcadores hemolíticos y parcial para valores de Hb > 100 g/L o incremento de 20 g/L y reducción de los marcadores hemolíticos sin necesidad de transfusión.7
Reticulocitos
Los reticulocitos son los precursores directos de los ER y solo representan aproximadamente 1 % de los ER en sangre periférica, aunque el intervalo de referencia puede variar entre laboratorios. Presentan un volumen corpuscular medio aumentado y un citoplasma basofílico producto de las trazas de ARN ribosomal.7
Los reticulocitos representan el índice de actividad hematopoyética de la médula ósea, generalmente están incrementados en las hemólisis y en otras condiciones patológicas y fisiológicas como en la hemorragia del embarazo, parto y en la aclimatación a las alturas. Aunque, en condiciones de hemólisis, la reticulocitosis compensatoria puede ser inadecuada o ausente en presencia de factores concomitantes que involucren daños a la médula ósea, tales como: enfermedades oncohematológicas, diseritropoyesis, deficiencias de hierro y vitaminas, infecciones o reacciones autoinmunes contra precursores hematopoyéticos. Este último es de particular interés en las AHAI, donde se reporta reticulocitopenia en 39 % de los niños y en 20 % de los adultos.2,8
La reticulocitopenia puede representar una emergencia clínica con altos requerimientos transfusionales y pobres resultados como lo observado por Brodsky en casos de HPN con anemia refractaria muy grave y en casos de AHAI fatal.9,10
Por esta razón los reticulocitos pueden ser evaluados como número absoluto o índice de respuesta de la médula ósea (BMRI, del inglés bone marrow responsiveness index):BMRI = [conteo absoluto de reticulocitos del paciente x (Hb del paciente/Hb normal)]. El valor de corte es < 121 y es útil para discriminar la hemólisis con inadecuada reticulocitopenia concomitante; con una sensibilidad del 90 % y especificidad del 65 %.11
La reticulocitosis es un importante marcador de recuperación de la hemólisis y de respuesta al tratamiento, que requiere, de 3 a 5 días junto con la administración de suplementos de folato, vitamina B12 y hierro en pacientes deficientes. En las AHAI, los reticulocitos permanecen elevados hasta que recuperen los valores de Hb. En pacientes con inadecuada reticulocitosis la eritropoyetina, permite corregir la anemia y reducir o evitar la hemólisis relacionada con la hipertransfusión como lo observado con agonistas de la trombopoyetina en la trombocitopenia inmune primaria.12
En enfermedades hemolíticas crónicas congénitas, los reticulocitos están ligeramente aumentados pero pueden elevarse dramáticamente en crisis hemolíticas agudas. En la esferocitosis hereditaria, los conteos globales de células se encuentran significativamente disminuidos después de esplenectomía, coherente con la reducción del proceso hemolítico a diferencia del déficit de piruvato-kinasa donde persisten conteos elevados después de este proceder.13,14
En pacientes con HPN bajo tratamiento, los conteos permanecen elevados por la persistencia de hemólisis por deposición del fragmento C3dg sobre los eritrocitos.15) En pacientes con válvulas cardíacas artificiales no existen cambios en los conteos. Bajo esta condición existe una hemólisis subclínica con niveles de Hb normal o ligeramente disminuidos.16
Esquistocitos
Los esquistocitos son fragmentos de los ER, visibles en lámina periférica como cuerpos irregulares. Derivan de la fragmentación mecánica de estas células producidas por vesículas como por ejemplo coágulos de fibrina, válvulas cardíacas artificiales u otro dispositivo intravascular.
En individuos sanos, los conteos de estos fragmentos son inferiores al 0,5 %. Conteos entre 3-10 % son típicos de la púrpura trombótica trombocitopénica (PTT), mientras que valores entre 0,5-1 % son indicativos de coagulación intravascular diseminada (CID). Una vez que los dispositivos intravasculares y la CID son descartados, el diagnóstico diferencial abarca las microangiopatías trombóticas causada por la activación de la hemostasia en pequeñas vesículas con consumo de plaquetas, factores de la coagulación y eritrocitos.17
En la PTT ocurre una hemostasia aberrante causada por el déficit congénito o adquirido de ADAMTS 13, una metaloproteinasa responsable de la degradación de los multímeros del factor von Willebrand. Otras causas de microangiopatias trombóticas son el síndrome hemolítico urémico (SHU) y el SHU atípico causado por la actividad de la toxina Shiga-like, la cual activa el sistema de complemento. En mujeres embarazadas con anemia hemolítica, la presión arterial, los conteos de plaquetas y las enzimas hepáticas deben ser cuidadosamente evaluadas, previendo la posible ocurrencia del síndrome de hemólisis con elevación de las enzimas hepáticas y bajos niveles de plaquetas (HELLP, del inglés hemolysis with elevation of liver enzymes and low platelets).18,19
Otros hallazgos importantes para realizar el diagnóstico diferencial son la proteinuria y el daño renal (más pronunciado en HELLP, en el SHU típico y atípico), actividad no detectable de ADAMTS 13 y síntomas neurológicos (común en PTT) e historia de diarreas producidas por E. coli (SHU atípico). Si se sospecha anemia hemolítica microangiopática, el examen de la lámina periférica permite un rápido diagnóstico. El tratamiento temprano de estas enfermedades dentro de las primeras horas del diagnóstico permite disminuir dramáticamente la mortalidad por estas causas.20
Deshidrogenasa láctica
La deshidrogenasa láctica (LDH) es la enzima que cataliza la conversión del lactato a ácido pirúvico y se localiza en el citoplasma de las células de varios órganos y tejidos, por ejemplo: corazón, músculo, hígado y cerebro. En el suero están presentes 5 isoenzimas que pueden ser producto del recambio celular fisiológico. Las isoenzimas LDH-1 y 2 están en los eritrocitos, que en condiciones hemolíticas se encuentran aumentadas, siendo una herramienta útil para distinguir la hemólisis extravascular de la intravascular. En la primera, estas se encuentran ligeramente aumentadas (AHAI por anticuerpos calientes y en las AHH) mientras que en la hemólisis intravascular se encuentran aumentadas 4 - 5 veces al límite superior del rango de referencia (en la HPN).
En pacientes con AHAI los niveles elevados de LDH son observados en el tipo caliente y en las formas frías con un rango térmico próximo a 37 oC. En la hemolisis intravascular causada por la activación del complemento, los valores elevados de esta enzima se correlacionan con la gravedad clínica y eventos trombóticos. En pacientes con infecciones sistémicas, la LDH se encuentra significativamente aumentada, sobre todo en aquellos con AHH.4
La LDH es útil en la evaluación de la respuesta al tratamiento ya que sus niveles disminuyen con la reducción de la tasa de hemólisis. Esto ha sido descrito en la AHAI y en el SHU típico y atípico posterior a la terapia y en la anemia hemolítica microangiopática después del recambio plasmático.21
En pacientes con HPN y bajo tratamiento con eculizumab, los síntomas provocados por la hemólisis regresan entre el primer o segundo día antes de la próxima dosis del medicamento y se asocian al aumento de los niveles de LDH, lo que sugiere que se acorte el intervalo entre la administración de las dosis o que aumente el volumen de medicamento.15
La LDH no solo aumenta en condiciones de hemólisis, sino también en la necrosis celular y en el daño tisular (infarto de miocardio, fallo cardíaco, en todas las formas etiológicas de hepatitis, en el esfuerzo muscular extremo y en tumores sólidos y hematológicos). Muy recientemente, la tasa LDH: aspartato-aminotransferasa de 22:12 ha permitido diferenciar la PTT de otras anemias hemolíticas microangiopáticas (SHU y HELLP), aún antes de los resultados del ensayo de actividad de la ADAMTS 13.22 Además, los niveles de la LDH pueden estar marcadamente elevados en pacientes con déficit de vitamina B12 y ácido fólico, por la eritropoyesis ineficaz y la destrucción prematura de los eritrocitos.23
Haptoglobina
La haptoglobina (Hp) es una glucoproteína sintetizada por el hígado y ubicada dentro de la fracción (2-globulina. Presenta propiedades antioxidantes e inmunomoduladoras y actúa como receptor de Hb libre por su unión estable a la Hb liberada producto de la hemólisis o del ciclo normal de los eritrocitos. Estos complejos Hp-Hb son rápidamente aclarados de la circulación por el sistema retículo endotelial vía receptor CD163, lo que previene la generación de especies reactivas del oxígeno y del daño renal. Después de la endocitosis, los complejos Hp-Hb son degradados en los lisosomas con la consiguiente disminución de la Hp.24
La Hp no es una proteína homogénea pues presenta dos alelos comunes llamados Hp1 y Hp2, lo que produce tres posibles variantes (1-1, 2-2, y 1-2) y polímeros de diferentes pesos moleculares distinguibles por electroforesis de alta resolución y diferentes comportamientos contra el estrés oxidativo.25
Estas tres variantes de Hp sufren una significativa reducción durante la hemólisis intravascular producido por el desbalance entre la Hb libre y los complejos Hb-Hp. En esta forma de hemólisis existe una pobre hemolisis extravascular de eritrocitos alterados estructuralmente escapados del aclaramiento por el sistema retículoendotelial. En la AHAI la Hp constituye el marcador más sensible de hemólisis y es el último en normalizarse después de la recuperación del paciente, permaneciendo disminuida aún en presencia de niveles normales de Hb.4
En el diagnóstico diferencial, los niveles reducidos de Hp, además de la hemólisis, se observan en el daño hepático, la malnutrición y en la haptoglobulinemia congénita. Por otro lado, sus niveles se encuentran aumentados en enfermedades inflamatorias, en fumadores y en el síndrome nefrótico, posiblemente enmascarando el proceso hemolítico.26
Bilirrubina
La bilirrubina IXa deriva del catabolismo del grupo hemo del anillo de protoporfirina IX, el grupo prostético de las proteínas involucradas en el transporte de O2 por la oxigenasa microsomal (Hb, mioglobina y citocromo P450).
El 85 % de la bilirrubina circulatoria deriva del catabolismo de la hemoglobina en órganos retículo endoteliales. Una eritropoyesis inefectiva constituye otra fuente adicional de bilirrubina. La bilirrubina es un buen marcador de hemólisis extravascular, y en menor medida, de hemólisis intravascular, donde una pequeña fracción de hemo liberado se une a la hemopexina y se cataboliza en el hígado. La bilirrubina producida en periferia es transportada al hígado, unida a la albúmina (bilirrubina no conjugada). En este órgano es convertida en bilirrubin-mono y diglucoronido por la enzima microsomal bilirrubin-UDP-glucoroniltransferasa y luego es excretada en la bilis (bilirrubina conjugada). Niveles altos de bilirrubina pueden ser el producto de un catabolismo aumentado de Hb (principalmente bilirrubinemia no conjugada) o disminución del aclaramiento hepático (bilirrubina conjugada).27
La hiperbilirrubinemia durante los procesos hemolíticos, usualmente no es superior a 4 mg/Dl; valores superiores indican una reducción en la función hepática, por lo cual se sugiere investigar los marcadores hepáticos. Valores de bilirrubina no conjugada superiores a 4 mg/dL son observados en la hemólisis masiva aguda (deficiencia de G6PD y en reacciones transfusionales)27 y en casos en que coexiste el síndrome de Gilbert, reportado en el 5 % de la población general y caracterizado por la reducción de la enzima bilirrubin-UDP-glucoroniltransferasa.28
La bilirrubina es un marcador de respuesta temprana al tratamiento a las 4 horas posteriores, una vez que cesa la hemólisis. En las AHAI la disminución de la bilirrubina no conjugada es observada alrededor del séptimo día posterior al inicio a la terapia con esteroides, evidenciando la respuesta al tratamiento.29 Además, se observan niveles disminuidos de bilirrubina no conjugada después de esplenectomía en la esferocitosis hereditaria30 y la bilirrubina circulatoria total tiende a la reducción en pacientes con HPN después del tratamiento con eculizumab.31
Ferritina
La ferritina es una proteína que almacena hierro y lo libera en situaciones de deficiencia. Está incrementada en varias condiciones hemolíticas crónicas como: defectos congénitos de las proteínas de membrana y enzimas eritrocitarias, en el síndrome de aglutininas frías crónica y en la anemia diseritropoyética congénita.11 Los pacientes afectados de HPN pueden presentar también este aumento en los valores de ferritina cuando son tratados con eculizumab, probablemente producto de la continua hemólisis o la reducción de sus niveles producto de la hemosiderinuria o por la pérdida de hierro.4
La ferritina es una proteína de fase aguda y puede encontrase aumentada por varias condiciones como en enfermedades metabólicas e inflamatorias (infecciones agudas y crónicas, hepatitis y tumores). La coexistencia de estas situaciones junto a la hemólisis crónica produce valores particularmente altos, al igual que en pacientes afectados de hemocromatosis hereditaria causada por la hemólisis crónica.32
Hemosiderinuria
Se denomina hemosiderinuria a la presencia de hemosiderina unida al hierro en la orina, lo que da una coloración oscura a esta. Típicamente se asocia a un patrón de hemólisis intravascular. La Hb liberada a la circulación excede la capacidad de unión de la Hp, es filtrada por el riñón y reabsorbida en el túbulo contorneado proximal. Aquí una porción de hierro es removida y almacenada como ferritina o hemosiderina para luego ser excretada en la orina. La hemosiderinuria es típica de la HPN, las reacciones transfusionales incompatibles, la deficiencia de G6PD, las quemaduras graves y las infecciones. Puede persistir por varias semanas después del cese de la hemólisis, mientras que la hemoglobinuria desaparece rápidamente.33
Alteraciones de otros tipos celulares
Se debe tener presente la posible reducción de los conteos de plaquetas y leucocitos en las deficiencias de vitamina B12 y ácido fólico. Además, la presencia de trombocitopenia sugiere la posibilidad de microangiopatía trombótica o síndrome de Evans. Este último puede ser reconocido rápidamente y tratado, constituye un factor pronóstico negativo en pacientes con AHAI.34
En la HPN los leucocitos y plaquetas pueden estar ligeramente reducidos y presentar un número deficiente de las moléculas CD55 y CD59 y ser destruidas por la activación del sistema del complemento. Mientras que la leucopenia y trombocitopenia muy grave se observa cuando la HPN tiene asociado fallo medular. Por otra parte, en pacientes con membranopatías y enzimopatías, la trombocitopenia ligera puede estar relacionada al hiperesplenismo y la trombocitosis se observa después de practicada la esplenectomía. A su vez, los dispositivos intravasculares pueden destruir, no solo a los ER, sino también a los leucocitos y plaquetas.35
Los parámetros hemolíticos pueden estar diferencialmente alterados en varias condiciones lo cual ayuda en la realización del diagnóstico diferencial de las anemias hemolíticas (según se mostró en la tabla). Sin embargo, muchos factores pueden complicar y enmascarar el cuadro clínico, lo cual evidencia la necesidad de una evaluación clínica y de laboratorio comprensiva. Por último, estos marcadores permiten monitorear la eficacia de los tratamientos.

