










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La anemia de Fanconi (AF) es un síndrome de inestabilidad cromosómica caracterizado, generalmente, por una combinación de rasgos dismórficos (baja estatura, microftalmia, hiperpigmentación de la piel, malformaciones cardíacas, renales y genitourinarias entre otras).
La mayoría de los pacientes presentan en el transcurso de la enfermedad, fallas a nivel de la médula ósea y alto riesgo de desarrollar tumores malignos.1,2 Los pacientes con síndromes por fallas en la médula ósea (SFMO) que tienen subyacente una AF no diagnosticada, no responderán a la terapia inmunosupresora que reciben usualmente los pacientes con anemia aplásica idiopática. Además, debido a la hipersensibilidad a los agentes quimioterapéuticos, los pacientes podrían morir debido a la toxicidad de estas drogas si se utilizan en dosis convencionales, por lo que un diagnóstico temprano de AF es esencial.3
La enfermedad presenta heterogeneidad genética y fenotípica debido a la existencia de varios subtipos genéticos. La identificación de estos se basa en el análisis de complementación o corrección del defecto molecular de las células del paciente con líneas celulares de referencia de AF, en las que se conoce el gen mutado para un grupo de complementación determinado. Si la deficiencia para reparar el daño del ADN inducido por agentes de entrecruzamiento no se corrige en las células del paciente, se concluye que pertenece a ese grupo de complementación particular.
Cada grupo de complementación, está definido por mutaciones en ambos alelos de un gen específico de AF. Por lo que, esta enfermedad presenta herencia autosómica recesiva en todos los subtipos a excepción del FANCB (grupo de complementación B de AF), ligado al cromosoma X, y del FANCR (grupo de complementación R de AF), cuya mutación causa una forma autosómica dominante de la enfermedad.1,2,4,5
Hasta el momento, 21 genes están implicados en la patogénesis molecular de esta enfermedad, designados con el prefijo FANC y letras seriadas del alfabeto:6FANCA (#227650), FANCB (#300515), FANCC (#227645), FANCD1 (#605724), FANCD2 (#613984), FANCE (#613976), FANCF (#613897), FANCG (#614082), FANCI (#609053), FANCJ (#609054), FANCL (#614083), FANCM (#609644), FANCN (#610832), FANCO (#613390), FANCP (#613951), FANCQ (#615272), FANCR (#617244), FANCS (#617883), FANCT (#616435), FANCU (#617247), FANCV (#617243), (respectivamente, se corresponden con: gen del grupo de complementación A, B, C, D1, D2, E, F, G, I, J, L, M, N, O, P, Q, R, S, T, U, V de AF). Se debe puntualizar que, de acuerdo con la base de datos en línea de herencia mendeliana en el hombre u OMIM (del inglés: Online Mendelian Inheritance in Man) el paciente reportado con FANCM afectado, fue encontrado posteriormente que tenía FANCA mutado, por lo que fue clasificado con FANCA. Los hallazgos hasta el presente no han sido concluyentes en relación con la hipótesis de que FANCM esté asociado a la AF.7
Esta base incluye, además, el gen FANCW (#617784), reportado por Knies y otros.6 Mutaciones en cualquiera de estos genes, no solo afectan la función canónica que tienen en la reparación del ADN y en el mantenimiento de la estabilidad del genoma, sino también otras funciones relacionadas con la respuesta protectora contra el estrés oxidativo, la inflamación, los aldehídos endógenos, la mitofagia y la virofagia. De modo que, la pérdida de la función de alguna de las proteínas en la AF, al ser multifuncionales, podría contribuir a la progresión de la enfermedad y a la variabilidad clínica de los pacientes, más allá de la alteración de la vía de reparación del ADN.4,5
Las manifestaciones clínicas en la AF son muy variables, algunos de los síntomas pueden solaparse con los observados en otros síndromes de inestabilidad genómica (síndrome de Bloom, ataxia telangectasia, síndrome de Nijmegen, síndrome de Seckel) o no presentar ninguno.1) Este fenotipo tan variable hace que el diagnóstico exacto sobre la base de las manifestaciones clínicas sea muy difícil.2,3,8,9
Las células derivadas de pacientes con AF deben presentar, de forma inequívoca, hipersensibilidad a las roturas cromosómicas inducidas por agentes de entrecruzamiento de ADN, tales como mitomicina C (MMC), diepoxibutano (DEB) o cisplatino, lo que constituye un rasgo distintivo de carácter diagnóstico en estos pacientes.2,3,9,10
El objetivo de este trabajo consistió en realizar el análisis discriminante de roturas cromosómicas inducidas por MMC en linfocitos de sangre periférica de pacientes cubanos con sospecha de AF.
Métodos
Se realizó estudio cualitativo de comparación de proporciones, desde mayo 2013 hasta octubre 2016. Fueron incluidos en este estudio 32 pacientes (11,4 ± 5 años) procedentes del Instituto de Hematología e Inmunología “Dr José Manuel Ballester Santovenia” y de la Red Nacional de Genética Médica.
Los pacientes fueron seleccionados tomando en consideración los criterios clínicos de la AF, fundamentados en la historia natural de la enfermedad, el examen físico, y asentados en la historia clínico-genética. Treinta pacientes presentaron SFMO y dos exhibieron rasgos dismórficos asociados a AF, pero sin fallas en la médula ósea. Además, se incluyeron 32 sujetos aparentemente sanos que actuaron como controles, en los que no se tuvo en cuenta la edad. Para la selección de estos últimos se precisó que no fueran hermanos de los pacientes y que no tuvieran antecedentes de síndrome de rupturas cromosómicas o de exposición a quimioterapia o radioterapia en el último mes.
En ambos grupos se aplicó un cuestionario para recolectar información relacionada con su historia clínica y la exposición a agentes químicos y físicos. Se colectaron 5 mL de sangre venosa en jeringuilla heparinizada.
Los pacientes, los padres de menores y los controles, dieron su consentimiento para participar en el estudio y sus datos fueron manejados confidencialmente. Se obtuvo la aprobación del comité de ética del Centro Nacional de Genética Médica (CNGM).
Se realizó el análisis de rupturas cromosómicas en linfocitos T de muestras de sangre venosa periférica cultivada en presencia de MMC y se siguió un protocolo semejante al descrito por Oostra y otros.9
Los cultivos se realizaron por 72 horas en medio Quantum PBL (Capricorn), que contiene fitohemaglutinina (PHA), a 37 ºC en ambiente de 5 % de CO2, acorde a protocolos estándares del laboratorio de citogenética del CNGM. Se prepararon tres cultivos, tanto para el paciente como para el control sano. Uno se utilizó como control mediante la adición de 50 µL de NaCl 0,9 % y dos para verificar el efecto de dos concentraciones de MMC en inducir aberraciones cromosómicas. Se utilizaron 16.6 µL y 100 µL de una solución de trabajo de MMC 30 µM para obtener las concentraciones finales de 50 nM y 300 nM.9 El NaCl y la MMC se añadieron al inicio del cultivo.
En cada cultivo fue analizado un mínimo de 50 metafases (teñidas con Giemsa), en las que se registraron los tipos de roturas identificados, así como el número de células aberrantes y normales. Para la evaluación final, todas las aberraciones fueron convertidas en “eventos de roturas” los cuales representan el principal tipo de aberración en las células características de la AF. Las roturas de cromátidas (gaps) y las roturas cromosómicas, fueron contadas como roturas simples; y las roturas tri- y cuatrirradiales como eventos de dos roturas cada uno. En otras figuras de intercambio el número de centrómeros es añadido al número de gaps y roturas abiertas.
Para la definición de un paciente con AF, se consideró la identificación de células típicas con 10 o más roturas por células. Así, a 50 nM de MMC una proporción notable de células debe mostrar roturas cromosómicas en la AF, mientras que a la concentración de 300 nM de MMC todas estarán afectadas por aberraciones cromosómicas y en la mayoría se observarán 10 o más roturas por células. Contrariamente, las células del sujeto control no deben mostrarse afectadas, excepto a 300 nM, donde pueden observarse hasta cinco roturas por célula. Para la evaluación de un paciente con AF en mosaico, se consideró la presencia de dos poblaciones de células con diferente sensibilidad a la MMC: una con células típicas con 10 o más roturas por célula, y otra con cinco o menos roturas por célula.
Las roturas cromosómicas inducidas por MMC en el paciente fueron comparadas con el cultivo basal (con NaCl) y el del control sano. La diferencia entre los cultivos de los pacientes y controles, tratados con cada una de las concentraciones de MMC se determinó por comparación de los porcentajes de metafases aberrantes mediante la prueba de chi-cuadrado.
Resultados
El estudio de roturas cromosómicas fue exitoso en 30 de los 32 pacientes. En seis (20 %) de los cultivos con resultados, el ensayo con MMC a 300 nM mostró un mayor número de roturas cromosómicas respecto a los controles, y esta diferencia fue significativa (p < 0,05); lo que permitió diagnosticarlos como AF. De estos seis solo dos manifestaban diversos rasgos dismórficos típicos de la entidad y los restantes presentaban únicamente anemia aplásica.
Por otra parte, dos pacientes presentaron poblaciones de linfocitos en las que la mayoría de las células (50-70 %) tratadas con 300 nM de MMC exhibieron un alto número de roturas e intercambios característicos de la AF, pero el resto de las células no mostró roturas cromosómicas. En estos dos hubo un aumento significativo de las roturas por célula con respecto a los controles (p < 0,05) y fueron diagnosticados como mosaicos para AF. Estos resultados son resumidos en la tabla, que representa la sensibilidad a las dos concentraciones ensayadas de MMC en el grupo control (n = 32), los pacientes no AF (n = 24) y pacientes AF detectados (n = 6). Se presenta la media y desviación estándar. Las frecuencias basales de rupturas cromosómicas por célula fueron ligeramente más altas en los 30 pacientes que en los controles, pero la diferencia no fue significativa (p > 0,005).
Tabla Porcentaje de roturas cromosómicas obtenidas en linfocitos T bajo la acción de la mitomicina C (MMC) en controles y pacientes
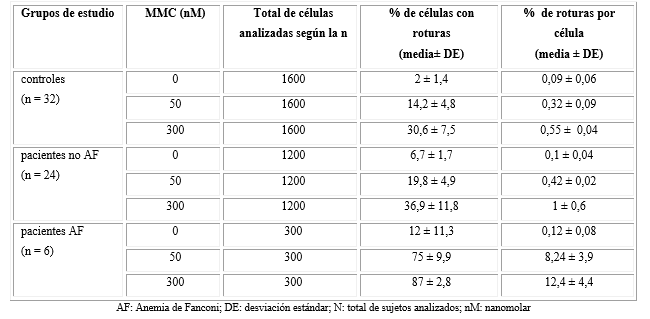
Las alteraciones cromosómicas inducidas por la acción de la MMC en los cultivos de linfocitos fueron superiores en los pacientes no AF que en los controles. Sin embargo, esta diferencia no fue significativa (p > 0,05) por lo que no hubo una distinción exacta entre los dos grupos. Como se presenta en la Fig., las roturas cromosómicas inducidas por MMC fueron dependientes de la concentración y existió una diferencia significativa entre la frecuencia de roturas a 50 y a 300 nM (p < 0,05) en los tres grupos.
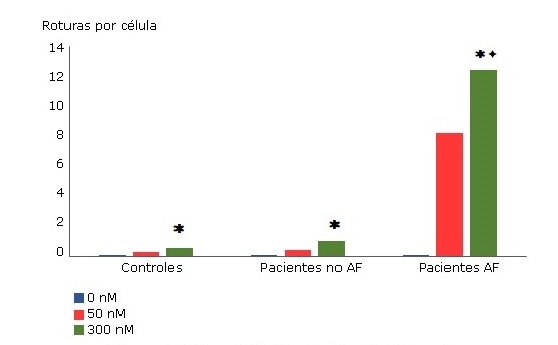 AF: anemia de Fanconi; MMC: mitomicina C; nM: namolar; Controles: 32 sujetos clínicamente sanos; Pacientes no AF: 24 pacientes con anemia aplásica; Pacientes AF: 6 pacientes con diagnóstico de anemia de Fanconi (: diferencias significativas (p < 0,05) entre la frecuencia de roturas a 50 y a 300 nM de MMC en los tres grupos. (: diferencias significativas (p < 0,05) en los pacientes AF a 300 nM en relación a los no AF y a los controles
AF: anemia de Fanconi; MMC: mitomicina C; nM: namolar; Controles: 32 sujetos clínicamente sanos; Pacientes no AF: 24 pacientes con anemia aplásica; Pacientes AF: 6 pacientes con diagnóstico de anemia de Fanconi (: diferencias significativas (p < 0,05) entre la frecuencia de roturas a 50 y a 300 nM de MMC en los tres grupos. (: diferencias significativas (p < 0,05) en los pacientes AF a 300 nM en relación a los no AF y a los controlesFig Medias de las roturas cromosómicas/células inducidas con el agente bifuncional alquilante mitimicina C en los linfocitos T de muestras de sangre cultivadas de los sujetos estudiados.
El ensayo con MMC reveló que a la concentración de 300 nM ocurre un aumento significativo, tanto de las roturas por célula como del porcentaje de células con roturas, entre pacientes con AF con respecto a los pacientes no AF y a los sujetos controles (p <0,05). Como muestra la Fig., las células típicas de AF con múltiples roturas (10 o más roturas /células) se observaron con los tratamientos de 50 nM y 300 nM de MMC, siendo mayor el nivel de roturas en este último.
Discusión
La inestabilidad cromosómica es un rasgo característico de un número de enfermedades humanas genéticamente conocidas como síndromes de rupturas cromosómicas. Algunas de estas incluyen: la AF, el síndrome de Bloom, la ataxia telangiectasia, y el síndrome de Nijmegen. En cada una de ellas, la inestabilidad ocurre en forma de un incremento de la frecuencia de roturas e intercambios que suceden tanto espontáneamente como después del tratamiento in vitro con varios agentes que dañan el ADN.11,12
La frecuencia basal media de roturas cromosómicas en las células de los sujetos controles en este estudio (0,09 roturas por célula) es comparable al dato que reporta el Registro Internacional de la la anemia de Fanconi de 0,25 roturas en células no tratadas (rango: 0,02 - 0,8).13 Este amplio rango de roturas cromosómicas espontáneas indica que la frecuencia de estos eventos en algunos pacientes con AF no difiere de la encontrada en individuos clínicamente sanos y por esta razón este parámetro no se utiliza con fines de diagnóstico.
En los cultivos de linfocitos T tratados con MMC de un paciente con AF es una regla común que una proporción sustancial de las células debe mostrar roturas cromosómicas en presencia de 50 nM de MMC,9) tal como se encontró en este estudio. A 300 nM de MMC no deben existir células sin daño y la mayoría debe estar en la categoría de 10 o más roturas/célula9) (más del 90 % en el estudio). En contraste, el cultivo celular de los controles sanos no debería estar afectado, excepto a 300 nM, donde por lo general, 30 % de las células debe mostrar de 1 a 5 roturas/célula,9) lo que se corroboró en los controles, con una media de 30,6 % de células con roturas cromosómicas en 50 células analizadas por muestra.
Dos pacientes presentaron una población mixta de linfocitos con hipersensibilidad y sensibilidad normal a la MMC. El mosaicismo en linfocitos se produce en una proporción considerable de pacientes con AF (estimado 25 al 30 %), y es causado por nuevas mutaciones compensatorias o reversión genética espontánea en el locus de la enfermedad en células progenitoras hematopoyéticas. Como consecuencia, se origina una población de células con una capacidad funcional de reparación del ADN, que son resistentes a los agentes clastogénicos, lo que se manifiesta con una sensibilidad disminuida a estos. Es decir, las células revertidas pueden corregir (parcialmente) la insuficiencia de la médula ósea. En estos casos se requiere, por tanto, realizar una segunda prueba de sensibilidad a estos agentes en cultivos de fibroblastos para demostrar los hallazgos característicos de las células típicas de la AF y corroborar el diagnóstico. El pronóstico clínico de estos pacientes es incierto, algunos sobreviven décadas sin fallo de la médula ósea o el surgimiento de procesos malignos hematológicos, mientras que otros sí desarrollan estas alteraciones a pesar de la presencia de poblaciones celulares revertidas.14,15,16
Debido al grado variable de reversión que puede incluir a células progenitoras pluripotenciales y a uno o varios linajes celulares, sería recomendable realizar un seguimiento a largo plazo de evaluación de la sensibilidad a MMC en linfocitos o fibroblastos.16,17
Giampietro y otros, reportan un 25 - 30% de pacientes sin rasgos dismórficos atribuibles a la AF, que fueron diagnosticados como tal, después de la aparición de anormalidades hematológicas.10
En pacientes sin anomalías congénitas es posible que la presencia muy temprana de distintos tipos de cáncer como la leucemia mieloide aguda y el cáncer de células escamosas de cabeza y cuello, entre otros, sea el primer indicio de AF. En tales casos es necesario comprobar el diagnóstico de AF antes de establecer los esquemas estándares de radio y quimioterapia, ante la posibilidad de desarrollar toxicidad letal debido al defecto en la reparación del ADN.2,17
En el presente estudio, cuatro pacientes con anemia aplásica, sin rasgos dismórficos obvios característicos de la enfermedad, resultaron tener AF. Otros dos pacientes diagnosticados como AF sin fallas en la médula ósea, pero con rasgos dismórficos pudieron ser detectados. Cabe destacar que todos fueron diagnosticados antes de la aparición de procesos malignos hematológicos. La anemia aplásica presente en 24 pacientes, con ningunas o pocas roturas cromosómicas inducidas, podría ser debido a otros síndromes de fallas en la médula ósea,3,9,17) de causa inmunológica18 o por deficiencias nutricionales, entre otras.19)
En la actualidad el trasplante de células madres hematopoyéticas o de médula ósea, es el único tratamiento curativo para las complicaciones hematológicas que se producen como consecuencia del fallo de la médula ósea, aunque no curan las manifestaciones no hematopoyéticas. Los trasplantes de donantes hermanos con antígenos de leucocitos humanos (HLA, del inglés: human leukocyte antigen) idénticos están asociados a excelentes resultados, con una supervivencia de 85 % para niños menores de 10 años y 65 % para niños y adultos combinados. En contraste, los trasplantes de donantes alternativos (emparentados o no) HLA compatibles son más complejos debido a un riesgo inmunológico incrementado.5,20) No obstante, los avances en este campo permiten que una mejor selección del donante, según edad, sexo y con una tipificación más precisa de alelos HLA, así como la aplicación de regímenes de acondicionamientos pretrasplante optimizados y de terapias inmunosupresoras posteriores al trasplante, garantizan que un trasplante de donante no emparentado HLA compatible tenga una supervivencia similar a la observada con uno de hermano donante HLA compatible.20
Un diagnóstico temprano en pacientes con AF (es decir, antes de la aparición de anormalidades hematológicas) podría proporcionar más tiempo para encontrar un donante HLA compatible, adecuado para el trasplante de médula ósea.10,15,20
Además, las familias en situación de riesgo (con un niño afectado) deben ser identificadas tempranamente y ofrecerles asesoramiento genético y posibilidad del diagnóstico prenatal, dado que la AF es, principalmente, un trastorno autosómico recesivo con un riesgo de recurrencia del 25 %.20,21 El retraso en la identificación de pacientes con AF y de familias en situación de riesgo se puede evitar mediante la realización de la prueba de daño en el ADN inducido por MMC en pacientes con macrocitosis y disminución de las plaquetas.21,22,23
El ensayo de roturas cromosómicas en cultivo de linfocitos frente a los agentes MMC o DEB es utilizado habitualmente como estrategia general para la detección de pacientes con AF, pre-anemia y anemia aplásica, con o sin rasgos dismórficos.2,3,9,17 Aunque se ha planteado que el ensayo con DEB se prefiere debido a posibles falsos positivos o negativos con otros agentes,3 en este estudio, el primero de su tipo en Cuba, el ensayo de rupturas cromosómicas inducidas por MMC resultó ser una herramienta útil para el diagnóstico anticipado de pacientes homocigotos con AF con rasgos dismórficos atribuibles a la enfermedad, pero sin manifestar aún anomalías hematológicas.

