










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Las inmunodeficiencias primarias (IDP) son enfermedades de origen genético causadas por alteraciones cuantitativas, funcionales, o ambas, de los mecanismos de la respuesta inmune.1,2,3,4
Se manifiestan por aumento de la susceptibilidad a infecciones, procesos autoinmunes, alergia y cáncer.1) El defecto molecular es conocido en la mayoría de ellas, pero se siguen describiendo nuevos genes cuyas mutaciones originan IDP y nuevos fenotipos clínicos.
Debido a la nomenclatura insuficiente de las IDP, la Organización Mundial de la Salud (OMS) convocó a los expertos para crear una clasificación coherente de estas enfermedades que se somete a revisión cada dos años con la finalidad de actualizarla, en 1996 esta recibió el aval de la Unión Internacional de Sociedades de Inmunología.5
La OMS reconoce más de 250 tipos de IDP. Se plantea que de 1/ 8 000 a 10 000 individuos tienen una IDP genética o congénita, pero existe un subregistro en la mayoría de los países ya que del 70 al 90 % permanecen sin diagnóstico.2
En Cuba se han diagnosticado IDP desde la década del 60 y la oficialización de la Inmunología como especialidad en 1976 contribuyó a ampliar el diagnóstico y seguimiento de estas enfermedades, posteriormente se creó el registro cubano de IDP y se han registrado en el país 337 pacientes.2
La incidencia de agranulocitosis es de 3,4 casos por millón de personas por año. Las inmunodeficiencias por deficiencias fagocíticas en número, función o ambos representan en Cuba el 12,2 %.2
La neutropenia se define como una disminución del recuento absoluto de neutrófilos (RAN), por debajo de las cifras normales según la edad y la etnia. Existen diversas clasificaciones de las neutropenias según la intensidad, la duración o la causa que las origina.
Según la intensidad se clasifican en: leves cuando el RAN oscila entre 1 000 y 1 500/mm3, moderadas entre 500 y 1 000/mm3, graves entre 200 y 500/mm3 y muy graves por debajo de 200/mm3.
Según la duración se clasifican en transitorias, cuando la duración es menor a 6 meses, o crónicas cuando es mayor. A su vez la neutropenia crónica puede ser permanente, intermitente con periodos de normalización espontánea y cíclica, con episodios de neutropenia que se repiten cada 21 días aproximadamente.
Según la causa, las neutropenias pueden ser clasificadas en alteraciones intrínsecas de la proliferación o maduración y ambas de las células mieloides en la médula ósea (neutropenias congénitas) (NC) y neutropenias secundarias, causadas por factores extrínsecos a las células mieloides (neutropenias adquiridas).6,7
El término NC hace referencia a la neutropenia crónica debido a un defecto genético constitucional, dentro de las cuales está el Síndrome de Kostmann y la neutropenia cíclica. Una definición más amplia incluye las NC que están asociadas con otras alteraciones.6,7,8
El cuadro clínico de la NC grave por alteración de la granulopoyesis fue descrito por el Dr. Rolf Kostmann, en una familia sueca, en la que los pacientes fallecían de infecciones graves en edades muy tempranas. Estos primeros casos seguían un patrón hereditario autosómico recesivo. Posteriormente se han descrito otros casos de NC grave con patrones hereditarios diferentes, autosómico dominante, autosómico recesivo, con patrón de herencia ligado al cromosoma X y casos esporádicos. Existe por tanto una heterogeneidad genética en el seno de las neutropenias congénitas.6,7
Los pacientes con NC grave desarrollan infecciones bacterianas graves dentro del primer año de vida. El riesgo de infección se correlaciona con el grado y duración de la neutropenia. Los sitios más afectados son la piel y mucosa de la orofaringe, asimismo, las infecciones pulmonares son frecuentes. Se presentan periodontitis, gingivitis y pérdidas dentales, aftas orales de mucosa y lengua, otra característica es la ausencia de infiltrados inflamatorios y formación de pus en la respuesta a infecciones bacterianas.7,8
Una gran variedad de bacterias Gram positivas y negativas se observan como agentes infecciosos, incluyendo especies Staphylococcus y Streptococcus, especies de Pseudomonas, infecciones fúngicas profundas por Aspergillus y Cándida son observadas en periodos prolongados de neutropenia. Síntomas gastrointestinales que asemejen una enfermedad inflamatoria intestinal, así como predisposición a fracturas patológicas debido a osteopenia.6,7
Los pacientes con NC padecen infecciones muy severas por lo que su diagnóstico y tratamiento se debe realizar precozmente, de no establecerse tratamiento oportuno de forma rápida la sobrevida de los pacientes se ve comprometida. El tratamiento definitivo es el trasplante de células progenitoras hematopoyéticas (TCPH), sin embargo existe mejoría con la administración de factor estimulante de colonias de granulocitos (G-CSF).7,8
Teniendo en cuenta el hecho de que se consideran raras estas enfermedades y la mala evolución que presentó el paciente, se decidió presentar a la comunidad científico-médica un caso de neutropenia congénita con evolución desfavorable.
Caso clínico
Lactante femenina, siete meses de edad, sin endogamia ni consanguinidad, producto de primera gestación, parto eutócico, a término de 40 semanas. Con padres sanos y sin hermanos. Lactancia materna exclusiva hasta los seis meses y esquema de vacunación no actualizado.
Antecedentes de síndrome purpúrico, trombocitocis, neutropenia, anemia severa y múltiples ingresos en terapia intensiva por neumonía, otitis media aguda, enfermedad diarreica aguda, celulitis periorbitaria, abscesos glúteos y en pabellones auriculares. El último ingreso por síndrome febril prolongado con varios abscesos en región glútea y en ambos pabellones auriculares, además de una bronconeumonía bilateral y síndrome de respuesta inflamatoria sistémica. Es valorada por Inmunología teniendo en cuenta la recurrencia y tórpida evolución de las infecciones.
Al examen físico se constató: mucosas hipocoloreadas, palidez cutánea mucosa. Se observaron lesiones de intertrigo moniliásico (placas eritematosas con borde activo y algunas lesiones exudativas), eritema perianal, lesiones abscedadas en ambos glúteos que comprometen casi su totalidad, además en ambos pabellones auriculares. Presentó abdomen globuloso, depresible, ligera hepatoesplenomegalia. En el sistema respiratorio, se apreció tiraje marcado supraclavicular e intercostal en ambos hemitórax. A la auscultación presentó disminución del murmullo vesicular y estertores crepitantes diseminados en ambos campos pulmonares con polipnea.
Se indicaron varios estudios complementarios, que se enumeran a continuación:
Exudado bacteriológico y micológico de lesiones de piel y partes blandas: Estafilococo aureus, Enterobater, Pseudomona sp, Cándida albicans.
Heces fecales y cultivos: Coco bacilo gram positivo y Blastocystis hominis.
Rayos X de tórax anteroposterior: Infiltrado intersticial bilateral en todo el pulmón, con opacidad en tercio inferior del pulmón derecho, no se aprecia área tímica.
Ultrasonido de Timo: No se visualiza área tímica.
Ultrasonido abdominal (3/1/2020): Riñones aumentados de tamaño, hepatomegalia de 4 cm, lesiones de aspecto inflamatorio de base derecha del pulmón, aumento de la ecogenicidad del tejido celular subcutáneo de toda la pared abdominal, más en el lado derecho, edematoso, no colección.
Ultrasonido de partes blandas (8/1/2020): Aumento de ecogenicidad en región perianal y en ambos glúteos, en el izquierdo, imagen de colección de 6 mm x 21 mm.
Tomografía Axial Computarizada (TAC) de abdomen (8/1/2020): Gran hepatomegalia que rebasó al cuadrante inferior derecho, en lóbulo derecho, área hipodensa de 51 mm x 55 mm, esplenomegalia de 79 mm x 43 mm, ambos riñones aumentados de tamaño.
Angio TAC (9/1/2020): Hepatomegalia, dos imágenes hipodensas, riñones aumentados de tamaño.
Cuantificación de inmunoglobulinas (Método: Nefelometría, equipo HITACHI COBAS C311)
Hemograma completo (Equipo: contador hematológico automatizado Mindray)
| Fecha | 03/01/2020 | 10/01/2020 | 15/01/2020 | Valores de Referencia |
|---|---|---|---|---|
| Leucocitos (x 109/L) | 15 | 5 | 5,4 | 4-10 |
| Polimorfonucleares (%) | 0,02 | 0,04 | 0,02 | 50-70 |
| Linfocitos (%) | 0,98 | 0,96 | 0,98 | 20-40 |
| Plaquetas (x 109/L) | 450 | - | 178 | 150-450 |
| Hematocrito | 0,21 | - | - | 0,37-0,54 |
| conteo absoluto de neutrófilos (mm3) | - | 205 | 108 | 2000-7500 |
Proteína C reactiva con valores que aumentaron en el tiempo en correspondencia con la evolución de las infecciones desde 297 mg/L hasta 548 mg/L.
Lámina periférica: Anisocitosis, hipercromía, leucopenia 3,7 x 109/L, plaquetas aumentadas en número 615 x 109L, macroplaquetas, neutropenia muy severa, linfocitosis.
Medulograma: Sistema megacariopoyético: hiperplástico; sistema granulopoyético: ausencia de elementos del gránulo, se observaron algunas células plasmáticas, abundantes linfocitos y sistema eritropoyético: íntegro.
La paciente fue valorada junto con la especialidad de Hematología, teniendo en cuenta los antecedentes, la clínica y los estudios se diagnosticó una IDP, específicamente una Neutropenia congénita. Se inició tratamiento con Hebertrans (1unidad), dosis 1 a 2 unidades por metro cuadrado de superficie corporal y se le administró 0,5 cc por vía subcutánea tres veces por semana. Leukocin 1bulbo subcutáneo 2 veces al día y Prednisona.
Se utilizó concentrado de granulocitos de donación paterna con escasa respuesta al tratamiento. También fue tratado con múltiples antimicrobianos (Ceftriaxona, Vancomicina, Azitromicina, Levofloxacino, Anfoterin B liposomal, Cotrimoxazol, Meronen, Clindamicina y Linezolid).
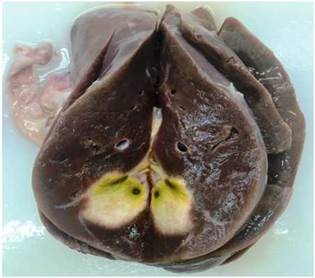
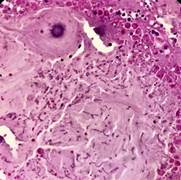
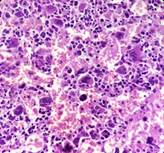
Evolucionó desfavorablemente y falleció por shock séptico. El informe de necropsia reflejó daño múltiple de órganos, sepsis generalizada por gérmenes oportunistas, agranulocitosis congénita, edema cerebral moderado, congestión vascular meníngea, neumonía necrotizante, micetomas en pulmón, hígado y estómago, hiperplasia y displasia megacariocítica, hipoplasia granulopoyética y sepsis generalizada de gérmenes como aspergillus, cándida, actinomices, datos que se corresponden con el diagnóstico de IDP (Figs. 1 , 2 y 3).
Discusión
La neutropenia congénita es una enfermedad poco frecuente y que representa una gran variedad de alteraciones. Aparece en edades precoces de la vida y debe tenerse en cuenta ante el diagnóstico de infecciones a repetición.
Los estudios de biología molecular describen mutaciones del gen de la elastasa 2 (ELA 2) como la causa más frecuente de NC, con un patrón de herencia autosómica dominante. En las formas recesivas se han identificado además de mutaciones en el gen HAX1, otras en los genes p14, G6PC3, TAZ y GASP. Sin embargo, en aproximadamente un 25 % de los casos no se puede identificar ninguna alteración genética, según los datos reportados por el Registro Internacional de NC.6,9
La presentación clínica inicial son las infecciones a repetición, sobre todo aquellas que aparecen durante los primeros meses de vida en forma de fiebre sin foco, infecciones cutáneas como abscesos e infecciones respiratorias. El diagnóstico de neutropenia de origen central se establece mediante el estudio de médula ósea, que muestra una detención en la maduración a nivel de promielocito/mielocito. Sin embargo, la existencia en algunos casos de una correlación entre genotipo y fenotipo sugiere la realización de un estudio genético dirigido de NC para tipificación de la misma.7,8,9
Cuba dispone de algunas técnicas de biología molecular para el diagnóstico de IDP pero no se realizan para la totalidad de estas enfermedades, así como el TCPH que aún no se ha aplicado en nuestro medio para el tratamiento de las mismas. El diagnóstico es clínico y de laboratorio con tratamiento sustitutivo con inmunoestimulantes.12
La paciente presentó todas las características descritas para este tipo de enfermedad, desde los antecedentes de infecciones a repetición con múltiples ingresos en cuidados intensivos, además de la presencia de microrganismos como hongos, bacterias y sobre todo la evolución desfavorable y poca respuesta al tratamiento, así como se encontró ausencia del área tímica y parámetros de la inmunoglobulina A en limites inferiores que refuerzan el diagnóstico de IDP.
El diagnóstico del caso de la paciente se correspondió con hallazgos clínicos descritos en la literatura, se evidencia en un estudio retrospectivo donde se incluyeron 109 niños diagnosticados con neutropenia y 4 presentaron NC con características similares a la paciente presentada.10
González y otros6 en un estudio retrospectivo sobre una serie de casos clínicos con 43 niños diagnosticados con neutropenia, encontraron neutropenias congénitas en 8 pacientes, de ellos se asociaron 3 a neutropenia benigna familiar, 2 con anemia de Fanconi y 3 a neutropenia cíclica, sin otros casos de neutropenia congénita grave, donde las infecciones recurrentes bacterianas, fueron las de mayor prevalencia lo que se relaciona con el caso descrito.
Cipe y otros11 reportaron un paciente en el que se encontró deficiencia del homólogo 1 de Jagunal (JAGN1), las cuales se definieron recientemente como defectos genéticos raros que causan NC grave. JAGN1 participa en la vía secretora y es necesario para la señalización mediada por el receptor del factor estimulante de colonias de granulocitos. Este gen es necesario para la ultraestructura normal y la granulación del retículo endoplásmico de las células progenitoras mieloides. Su defecto está relacionado con una mayor predisposición a la apoptosis. En nuestra paciente no se detectaron otras anomalías.
Otra investigación que se relaciona con la información obtenida del caso clínico presentado es la realizada con datos de 28 años en 11 pacientes pediátricos con NC, dónde la mediana de edad al diagnóstico fue de 4 meses, la clínica inicial en todos los casos fue infección grave. El aspirado de médula ósea mostró en todos los casos detención madurativo a nivel de promielocito. El estudio genético mostró en 3 mutaciones, 2 que afectaban al gen ELA2 y una en el gen G6PC3. El G-CSF fue el tratamiento de elección en 9 pacientes, 3 no respondieron a G-CSF, indicándose un TCPH alogénico. Contrasta que esta paciente no respondió al tratamiento con G-CSF.
Las inmunodeficiencias primarias son enfermedades de causa genética que involucran al sistema inmune. La presentación clínica es muy variable, desde pacientes casi asintomáticos hasta aquellos con manifestaciones muy graves. Se presentó una paciente con NC, confirmado por necropsia; la cual tenía múltiples infecciones con evolución desfavorable y no respuesta al tratamiento. El diagnóstico precoz es el elemento clave para la reducción de la morbilidad, mortalidad y mejoría de la calidad de vida de estos pacientes.

