










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Investigaciones Biomédicas
versión On-line ISSN 1561-3011
Rev Cubana Invest Bioméd v.18 n.2 Ciudad de la Habana may.-ago. 1999
Instituto de Ciencias Básicas y Preclínicas "Victoria de Girón"
La enfermedad por quemaduras como modelo de respuesta inflamatoria sistémica
Dr. Félix Broche Valle, Dra. Ela M. Céspedes Miranda, Dr. Alberto Saldaña Bernabeu y Dr. Arturo L. Cruz PérezRESUMEN
La quemadura corporal es una violenta agresión que modifica todos los mecanismos de la homeostasis orgánica y por su connotación clínica y social es un problema que enfrentan los servicios médicos en la sociedad contemporánea. Desde el punto de vista fisiopatológico, en el paciente quemado se desarrolla un Síndrome de Respuesta Inflamatoria Sistémica (SRIS) caracterizado por la hiperactivación de todos los mecanismos de defensa. La disregulación de estos mecanismos conduce al daño de los tejidos propios cuyas consecuencias se expresan en alteraciones morfofuncionales de todos los sistemas. Con independencia de la etiología, evolución inicial, manejo terapéutico y respuesta individual, la sepsis generalmente complica la evolución del gran quemado. En este trabajo, se presentan elementos clínicos e histopatológicos de la enfermedad por quemadura y se comentan los mecanismos moleculares de la respuesta inflamatoria sistémica destacando el papel de los mediadores de la comunicación intercelular.Descriptores DeCS: QUEMADURAS/patología; SINDROME SEPTICO/ /fisiopatología.
Aldrich en 1894 definió que la quemadura es una pérdida de sustancia de la superficie corporal por destrucción de la piel y el tejido subcutáneo ocasionada por alteraciones térmicas que comprenden el calor, el frío, agentes químicos, la electricidad y las radiaciones.1 Todos los tipos de quemaduras poseen un común denominador: la producción de alteraciones histológicas de la piel y la aparición de un síndrome de respuesta inflamatoria sistémica (SRIS).2
La piel es el tejido más extenso del organismo y diana primaria en la quemadura corporal. Fue considerada durante mucho tiempo como una estructura sólo de protección, pero se ha demostrado su carácter inmunocompetente.1-3
Los queratinocitos (cerca del 95 % de las células epidérmicas) pueden actuar como células presentadoras de antígeno y sintetizar mediadores de la comunicación intercelular que intervienen en el proceso inflamatorio. Por otra parte, las células de Langerhans (3-5 % de las células epidérmicas) funcionan como fagocitos tisulares así como en la presentación de antígenos a los linfocitos T CD4+.2 Los polimorfonucleares neutrófilos (PMN) circulantes son atraídos hacia la piel afectada y su hiperactivación contribuye a potenciar el daño generado por el trauma inicial.4
Cuando la extensión de la quemadura rebasa ciertos límites deja de ser un trastorno local para convertirse en la "enfermedad por quemadura"1 que requiere de un tratamiento intensivo. En este contexto, nuestro trabajo se orienta en función de profundizar en el estudio de los mecanismos moleculares involucrados en la fisiopatología de la enfermedad por quemaduras con la óptica del enfoque multidisciplinario que la complejidad del problema nos impone.
Consideraciones generales sobre la enfermedad por quemaduras
Al analizar la evolución del quemado debe distinguirse entre el efecto agudo de la quemadura que conduce al shock hipovolémico en las primeras 48-72 h y un efecto de evolución progresiva que se convierte en la "enfermedad del quemado".2La teoría del choque nervioso de Dupuyttren es la más antigua, se le atribuye al choque neurógeno por efecto del dolor el origen de todos los trastornos en el gran quemado. Eppinger sostuvo que las lesiones producidas por la quemadura debían atribuirse a trastornos de la permeabilidad capilar, en tanto Roplleer demostró en el suero de los quemados graves la existencia de una sustancia con tal efecto.1
Lorthioir señaló la liberación de enzimas proteolíticas intracelulares como causa de agresión generalizada a los tejidos. Otros autores identifican como causa fundamental de las alteraciones viscerales del gran quemado a la hipoxia hística.1,5
Hayde y Vogt sostuvieron el origen tóxico de los trastornos humorales, aunque sin precisar la naturaleza de las toxinas.1 Las toxinas del quemado pueden generarse a partir de la coagulación y destrucción hística producidas por el accidente, por la actividad metabólica de las células vecinas, en particular los fagocitos tisulares, el endotelio vascular y los fagocitos circulantes o los microorganismos que colonizan la piel o alcanzan la circulación sistémica.
Duval atribuyó la muerte de los quemados graves a una toxina polipeptídica producida por la propia quemadura.1 Se ha identificado un principio tóxico que puede ser separado por precipitación salina y es de naturaleza lipoproteica, lo cual sugiere su probable procedencia de las membranas celulares.2,6 Esta toxina está constituida por 6 subunidades asociadas a proteínas del shock térmico, pesa entre 4-16 kDa y el 40 % del peso total de la partícula corresponde a lípidos.2,7,8
Los fagocitos pueden activarse tras su exposición a este complejo lipoproteico (CLP). Sin embargo, aunque este complejo induce una activación inicial, algunos días después de la quemadura disminuye significativamente el tiempo de vida media y la capacidad de respuesta de todas las células fagocíticas.2 El CLP genera una respuesta inmune específica.2,9
En estudios realizados con inóculos de Pseudomona sp. en ratones se obtuvo el 20 % de mortalidad en tanto tras la administración conjunta con una dosis subletal del CLP se obtuvo el 80 % de mortalidad. Múltiples estudios demuestran que el conjunto de las diversas toxinas identificadas hasta el momento en pacientes quemados ejerce un efecto citotóxico mucho menos pronunciado que el CLP, especialmente sobre las células del sistema reticuloendotelial.2
Kosaka K y otros, identificaron un epóxido de linoleato sintetizado por los polimorfonucleares neutrófilos (PMN) cuya concentración exhibe una cinética bifásica con máximos durante y después de la primera semana que sigue a la quemadura. Se obtuvo una correlación positiva entre los niveles de esta leucotoxina y la superficie corporal dañada, la tasa de mortalidad y la concentración plasmática de proteína C reactiva.10
La producción de neopterina por los fagocitos también se incrementa, de inicio como indicador de la respuesta de fase aguda al trauma tisular, pero posteriormente es inducida por las endotoxinas bacterianas.11
En pacientes quemados con más del 20 % de superficie corporal comprometida, se elevan las concentraciones plasmáticas de endotelina 1 y trombomodulina, proporcionalmente con la concentración plasmática de factor de necrosis tumoral (TNF), la severidad del cuadro inflamatorio sistémico y la presencia de sepsis.12
En todos los quemados graves se incrementa la lipolisis y por tanto, la concentración de ácidos grasos libres, proceso estimulado por los glucocorticoides, interleuquina 1 (IL-1) y TNF. Sin embargo, IL-1 y TNF también estimulan la síntesis hepática de lipoproteínas de muy baja densidad; éstas pueden fijar e inactivar las endotoxinas bacterianas. Estos pacientes presentan también hipocetonemia.13
Desde el punto de vista histopatológico, en las quemaduras superficiales se produce vasodilatación cutánea con área de espongiosis en el epitelio, acantólisis secundaria que da lugar a la formación de flictenas y ampollas dermoepidérmicas con desprendimiento total de la epidermis y edema intenso de la dermis. En las quemaduras profundas en el nivel de la piel, es característica la escara constituida por los tejidos necróticos.1
Poco tiempo después de la quemadura, aparece vasodilatación y edema cerebral marcados con degeneración neuronal difusa. Las manifestaciones neurológicas iniciales del quemado incluyen inquietud y ligera desorientación. En los casos complicados aparecen progresivamente excitación, delirio y coma.1
En el miocardio aparecen áreas de necrosis, hemorragias focales, pericarditis fibrinosas y miolisis que clínicamente se expresan por arritmias e insuficiencia cardíaca.1,14
Las alteraciones de la integridad y permeabilidad vascular son evidentes en el nivel del foco de quemadura. En los casos más graves pueden aparecer focos de necrosis en la pared de los grandes vasos así como microtrombosis generalizada. El sistema hemolinfopoyético muestra alteraciones morfofuncionales en todos sus componentes en el nivel medular y periférico.1
En los túbulos renales aparecen depósitos de hemoglobina con degeneración parenquimatosa difusa y signos de fallo renal que puede llegar a la anuria. En los casos más graves aparece necrosis cortical bilateral, nefritis intersticial, abscesos múltiples e infartos difusos.1
Durante las primeras 48-72 h aparece edema pulmonar intenso con áreas de enfisema y neumonía reticulohiperplástica. Clínicamente estos pacientes presentan un cuadro de insuficiencia respiratoria progresiva. La congestión intensa, las hemorragias focales y las ulceraciones son manifestaciones frecuentes en la mucosa del tubo digestivo. Algunos casos presentan un síndrome ictérico.1
Los microorganismos causantes de la infección del quemado proceden al menos, de la piel, la mucosa intestinal1,5,15 y el medio ambiente.16,17 A menudo existen asociaciones de microorganismos multirresistentes. Las bacterias gramnegativas son la causa principal de la sepsis sistémica en los pacientes quemados (aproximadamente el 65 % de los casos) seguidas por las grampositivas (20 %), hongos, rickettsia y virus.18,19
En el nivel de la piel quemada es mucho mayor la incidencia de infecciones con respecto a otras lesiones traumáticas a causa de la pérdida de continuidad más o menos extensa de la barrera cutánea, primera línea de defensa del organismo, y a la presencia de alteraciones cuantitativas y cualitativas en la respuesta inmune humoral y celular de estos pacientes.20 Las quemaduras pueden presentar signos de infección local como: secreción verdosa fétida o escaras aparentemente secas por debajo de las cuales se acumula pus, en tanto en el nivel sistémico los signos clínicos de la infección local son la fiebre, leucocitosis, anemia progresiva, retraso o detención del proceso cicatrizal.1
La infección del quemado debe enfocarse como generalizada y tratarse local y generalmente. No obstante, la asociación microbiana por incorporación de nuevos gérmenes por vía externa o por la resistencia bacteriana a la antibioticoterapia complica notablemente el tratamiento.1
El paciente quemado pudo presentar: Bacteriemia y Endotoxemia (se refieren a la presencia de hemocultivos positivos y de endotoxina bacteriana en sangre respectivamente; Sepsis que implica la evidencia clínica de infección acompañada de reacción sistémica (taquipnea, taquicardia, leucocitosis, fiebre);18,21 Síndrome séptico que incluye sepsis con alteraciones en la perfusión de 1 o varios órganos y Shock séptico que requiere reposición de volumen y tratamiento vasopresor.18
Con el incremento de los niveles plasmáticos de endotoxinas bacterianas aumenta la incidencia de insuficiencia multiorgánica y la tasa de mortalidad; de hecho, la endotoxemia persistente es siempre un indicador de mal pronóstico para el paciente quemado.22
Las estrategias más recientes basadas en la atención integral del paciente, eliminación temprana de los tejidos necróticos, uso de poliquimioterapia antimicrobiana local y sistémica y la hospitalización en condiciones de aislamiento han mejorado el pronóstico general del gran quemado23 pero aún son muchos los problemas que requieren consideración y solución adecuada tanto en el orden de la investigación básica como en el campo de la práctica asistencial.
El síndrome de respuesta inflamatoria sistémica en la fisiopatología de la enfermedad por quemadura
Aunque en la reacción inflamatoria participan factores celulares y humorales en el nivel local y sistémico, el SRIS se reconoce como el conjunto de alteraciones morfofuncionales que se derivan de la hiperactivación prolongada de dichos factores con independencia de la fuente de daño primario.24 Bone C. definió el SRIS como la reacción inflamatoria masiva resultante de la liberación de mediadores sistémicos que conducen a la disfunción multiorgánica.25Los criterios de Bone para el diagnóstico de la respuesta inflamatoria sistémica de origen séptico son los siguientes:
- Fuente primaria de infección confirmada.
- Temperatura axilar < 36 o >38 E.
- Frecuencia cardíaca > 90 latidos/min.
- Frecuencia respiratoria > 20 resp./min.
- Leucograma:
> 10 % de células inmaduras.
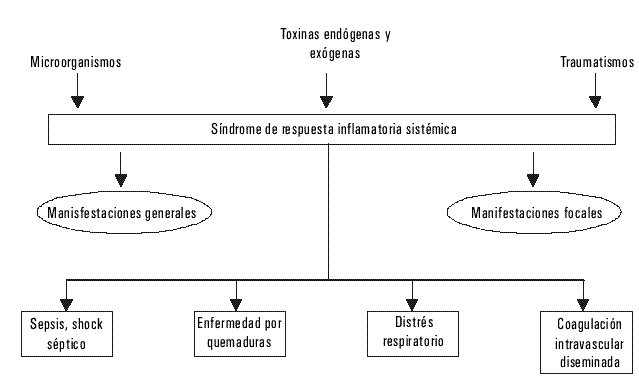
Fig. El síndrome de respuesta inflamatoria sistémica en la patología clínica.
La hiperactividad de los leucocitos, en particular los PMN y las células endoteliales constituye el elemento fisiopatológico esencial en el SRIS.24,27.28 La quimiotaxis, adherencia, migración a través del endotelio y actividad fagocítica son las principales funciones biológicas que expresan las células activadas en el curso de la reacción inflamatoria aguda.
Los macrófagos tisulares al actuar como células presentadoras de antígeno liberan factor de necrosis tumoral a (TNF a) e IL-1 que a su vez inducen por un mecanismo autocrino la síntesis de IL-6 e IL-8. Esta úlitma se comporta como potente activador de los PMN.29 Otros estímulos como el complemento activado (C5a), el factor activador plaquetario, leucotrieno B4, peróxidos lipídicos, inmunocomplejos y restos celulares inducen también la activación de los PMN.30
En todos los eventos del proceso inflamatorio interviene un complejo sistema de "señalización" integrado por medidoras de la comunicación intercelular. En 1974 Cohen utilizó el término "citoquinas" para identificar un grupo de polipéptidos con acciones locales y sistémicas, que participan en el control de diversas poblaciones celulares y que no derivan de los linfocitos. Actualmente, se define como citoquinas un grupo de proteínas inducibles que modulan la actividad (metabolismo, síntesis y secreción de otras proteínas, etc), proliferación y diferenciación celular.31
Según Bone C., en el desarrollo del SRIS pueden distinguirse 3 etapas de acuerdo con la actividad de las citoquinas involucradas en la respuesta:
Primera etapa que se caracteriza por la liberación local de citoquinas como respuesta al trauma y la infección.
Segunda etapa, donde se incrementa la concentración de citoquinas en la circulación sistémica.
Tercera etapa que se define por la reacción generalizada en la que la liberación masiva y prolongada de citoquinas conduce al daño vascular y de órganos diana.25 Entre las citoquinas proinflamatorias que desempeñan una función esencial en la respuesta aguda a la infección y el trauma tisular se incluyen, entre otras, IL-1, IL-6, IL-8 y TNF.26,31,32
Como IL-1 se conocen 2 proteínas (IL-1 a e IL-1 b) sintetizadas por monocitos, macrófagos, linfocitos, queratinocitos, células de Kupffer, hapatocitos, fibroblastos y células de la glia entre otras.33
Se conocen 2 tipos de receptores y sólo el tipo I (en linfocitos T, fibroblastos, queratinocitos y células endoteliales) puede iniciar el mecanismo de transducción de señales. Sus principales funciones incluyen la estimulación de su propia síntesis e inducción de la síntesis de TNF e IL-6 (por los monocitos y macrófagos) y de IL-2, IL-4 y factor estimulante de colonias de granulocitos/monocitos (por los linfocitos T). Estimula la proliferación y actividad de los linfocitos B, induce la síntesis de prostaglandinas en células endoteliales y del músculo liso, aumenta la síntesis de las proteínas de fase aguda en los hepatocitos e induce la síntesis de colagenasa en fibroblastos y mesotelio sinovial. También induce la fiebre.33
La IL-8 es sintetizada por monocitos/ /macrófagos, eosinófilos, linfocitos, neutrófilos, fibroblastos, células endoteliales, queratinocitos, hepatocitos, astrocitos y condrocitos. Sus principales acciones biológicas son la quimiotaxis, activación de leucocitos y expresión de varias moléculas de adhesión intercelular (MAI). Es además un factor angiogénico. La IL-8 actúa por unión a 3 receptores asociados a proteínas G en la membrana citoplasmática.34
El TNF a (caquectina) y el TNF b _(linfotoxina) son 2 proteínas con el 30 % de identidad secuencial, que se incluyen entre las citoquinas proinflamatorias por su papel central en la iniciación de la respuesta inmune frente a infecciones y algunos tipos de tumores. El TNF a es sintetizado por PMN, linfocitos activados, macrófagos, astrocitos, células endoteliales, células musculares lisas y algunas células transformadas. El TNF b es sintetizado sólo por linfocitos.35
Virtualmente todas las poblaciones celulares poseen al menos 1 de los 2 tipos de receptores para TNF. Al parecer el receptor tipo I está vinculado con la actividad lítica en tanto el tipo II regula la proliferación y activación linfocítica. Se han identificado proteínas solubles que forman complejos inactivos con el TNF.35
La concentración plasmática de TNF se eleva significativamente y de forma persistente en pacientes quemados que fallecen por síndrome séptico.36-38
Las MAI son proteínas de membrana que intervienen en la interacción y unión de células sanguíneas a sus similares, al endotelio y a la matriz subendotelial.39,40 La mayoría de las MAI conocidas se incluyen dentro de 4 superfamilias: inmunoglobulinas, integrinas, selectinas y cadherinas.40
La superfamilia de inmunoglobulinas incluye las MAI (ICAM-1, ICAM-2, ICAM-3, VCAM-1, MadCAM-1) que reconocen a las integrinas presentes en la membrana de los leucocitos y promueven la marginación y posterior extravasación de éstos a través del endotelio.39,40
La molécula de adhesión intercelular 1 (ICAM-1) es una licoproteína de membrana que se expresa en monocitos, células endoteliales, linfocitos, células dendríticas y epiteliales. Su función principal es la de intervenir como mediador de la adhesión leucocitaria al endotelio vascular activado.41
Las selectinas son una familia de glicoproteínas que se activan en las fases iniciales de la extravasación leucocitaria durante el proceso inflamatorio.39,40 En particular, la L-selectina interviene en el "reclutamiento" de los leucocitos circulantes, su adherencia al endotelio y extravasación39,42 hacia los sitios de inflamaciones aguda y crónica.
La L-selectina (constitutiva de los PMN) y las b 2 integrinas (inducibles durante el proceso inflamatorio) facilitan la adherencia de estas células a los endoteliocitos a través del reconocimiento de sus receptores (E-selectina para la L-selectina, ICAM-1 e ICAM-2 para las b 2 integrinas). Las células endoteliales activadas sintetizan también IL-8.43
Además de la hiperproducción de estas citoquinas, se ha reportado también un aumento asociado de la producción de IL-6 por los macrófagos, lo cual podría ser una de las causas de la respuesta celular deficiente en estos pacientes, aumentando su susceptibilidad a las infecciones.44 Las concentraciones plasmáticas de TNF, IL-6 e IL-8 alcanzan valores más altos en los quemados que desarrollan sepsis y en los que fallecen en insuficiencia mul-tiorgánica.45
Los fagocitos y el endotelio son los principales puntos de generación de ERO en el SRIS. En pacientes críticos se han encontrado altos niveles de lipoperóxidos y disminución de varios sistemas antioxidantes.46 La instalación de la insufiencia multiorgánica va precedida generalmente por un incremento en la producción de NO y ERO en el nivel de monocitos y macrófagos;47 evidencias que sugieren el papel del estrés oxidativo en la fisiopatología del gran quemado.
En términos generales, la magnitud de la lesión, rapidez de la terapia de reposición hidroelectrolítica y corrección de los desequilibrios acidobásicos, estado de salud anterior, procederes para la eliminación del tejido necrótico, el tratamiento local y sistémico así como la respuesta individual de cada organismo, influyen en la evolución del SRIS en el gran quemado. Pero indudablemente la sepsis es uno de los factores más importantes, pues impone la necesidad de hospitalización prolongada en unidades cerradas, a lo cual se asocia también el fenómeno de la resistencia a los antibióticos, entre otros factores.48
La consideración de los elementos comentados supone que los criterios de pronóstico, evolución y manejo de la conducta terapéutica pudieran complementarse con la inclusión de indicadores bioquímicos cuya base analítica se sustenta en la participación de los mediadores de la comunicación intercelular y el estrés oxidativo como eventos metabólicos de la respuesta inflamatoria sistémica del paciente quemado.
En una serie de artículos posteriores se abordarán en detalle los aspectos relacionados con la biología molecular de la respuesta inflamatoria sistémica y su aplicación al modelo de la enfermedad por quemaduras.
SUMMARY
A body burn is a violent agression that modifies all the mechanisms of organic homeostasis and is also a problem facing the medical services in the contemporary society because of its clinical and social connotation. From the physiopathological viewpoint, a burned patient develops a systemic inflammatory response characterizaed by hyperactivation of all its defense mechanisms. The de-regulation of such mechanisms leads to damage of tissues which is expressed as morpho-functional modifications of all systems. Regardless of the ethiology, initial evolution, therapeutic management and individual response, sepsis generally complicates the evolution of burn injuries. This paper sets forth the clinical and histopathological elements of burn-related diseases and comments on the molecular mechanisms of systemic inflammatory response, underlining the role of the intercellular signalling mediators.
Subject headings: BURN/pathologic; SEPSIS SYNDROME/physiopathology.
REFERENCIAS BIBLIOGRÁFICAS
- Kirschbaum MS. Quemaduras y cirugía plástica de sus secuelas. La Habana: Editorial Científico Técnica, 1987;4:129.
- Allgower M, Schoenenberger GA, Sparkes BG. Burning the largest immune organ burns 1995;21(Supp 1):537-47.
- Pejnovic N, Lilic D, Zunic G, Colic M, Kataranovski M, Dujic A. Aberrant levels of cytokines within wound after burn injury. Arch Surg 1995;130(9):999-1006.
- Schoenenberger GA, Baven UR, Cueni LB. Isolation and characterization of a cutaneous lipoprotein with lethal effects produced by thermal energy in mouse skin. Biochem Biophus Res Commun 1971;42:975-82.
- Garner WL, Rodríguez JL, Miller CG, Till GO, Rees RS, Smith DJ, Remick DG. Acute skin injury releases neutrophil chemoattractans. Surgery 1994;116(1):42-8.
- Schoenenberger GA, Cueni LB, Baven U. Isolation and characterization of a toxic lipid-protein complex formed in mouse skin by controlled thermal energy. Biochem Biophys Acta 1972;263:49-163.
- Baron P, Traber LD, Traber DL, Nguyen T, Hollyoak M, Heggers JP, Herndon DN. Gut failure and translocation following burn and sepsis. J Surg Res 1994;57(1):197-204.
- Allgower M, Cueni LB, Stadtler K. Burn toxin in mouse skin. J Trauma 1973;13:95-111.
- Teodorczyk-Injeyan J, Sparken BG, Mills GB. Impairment of T cell activation in Burned Patients: A possible mechanism of Thermal injury-induced immunosuppression. Clin Exp. Immunol 1986;65:570-87.
- Miles JM. Lipid fuel metabolism in health and disease. Curr Opin Gen Surg 1993;78:84.
- Varriale P, Ramaprasad S. Septic cardiomyopathy as a cause of long QT syndrome. J Electrocardiol 1995;28(4):327-29.
- Yang HM, Guo ZR, Sheno ZY. Delayed fluid resuscitation-induced bacterial translocation after lethal thermal injury. Role of oxygen free radical injury of intestinal mucosa. Chung Tlua I Hsueh Tsa Chih 1994; 74(9):552-84.
- Kosaka K, Suzuki K, Hayakawa M, Sugiyama S, Ozawa T. Leukotoxin, a linoleate epoxide: its implication in the late death of patients with extensive burns. Mol Cell Biohem 1997;139(2):141-8.
- Epstein MD. The Role of the gastrointestinal tract in the development of burn sepsis. Plast Reconstr Surg 1992;90:524-31.
- Yao YM, Yu Y, Wang YP, Tian HM, Sheng ZY. Elevated serum neopterin level: its relation to endotoxemia and sepsis in patients with major burns. Eur J Clin Invest 1996;26(3):224-30.
- Schiller HJ. Tissue perfusion in critical illnesses antioxidant Suppl):S92-102.
- Nakae H, Endo S, Inada K, Yamada Y, Takakuwa T, Yoshida M. Plasma levels of endothelin 1 and thrombomodulin in burn patients. Burns 1996;22(8):594-7.
- Páramo J, Rocha E. Nuevos conceptos sobre el papel de la hemostasia en la patogenia y tratamiento de la sepsis. Med Clin 1994;103:260-3.
- Thomas RM, Letulzo Y. Clinical and etiological diagnosis of septic states. Rev Prat 1993;43(1):35-40.
- Sanyal SC, Mokaddas EM, Gang RK, Bang RL. Microbiology of septicaemia in burn patients. Ann Burns & Fire Dis 1998;Vol XI(1):19-22.
- Natanson C. Selected treatment strategies for septic shock based on proposed mechanisms of pathogenesis. Ann Int Med 1994;Vol. 120(9):771-83.
- Weber JM, Tompkins DM, Improving Survival, infection control and burns. AACN-Clin Issues Crit Care Nurs 1993;4(2):414-23.
- Simms H, D'Amico R. Polymorphonuclear leukocyte dysregulation during the Systemic Inflammatory Response Syndrome. Blood 1994;Vol. 83(5):1398-407.
- Bone RC. Toward a theory regarding the pathogenesis of the Systemic Inflammatory Response Syndrome: What we do and what we do not know about cytokine regulation Crit Care Med 1996;24(1):163-72.
- Koichiro T, Shigeki M. Early events and stimulants triggering oxidative metabolism in neutrophils. En: Knox Van D, Castianova V, eds. Cellular Chemiluminiscence CRC Boca Ratón: Press Inc., 1982;1:113-27.
- R & D Systems. 1995 Catalog. cytokines and related reagents. Folleto comercial y promocional. R & D Systems Inc. minneapolis. USA, pág 3-5.
- Eduards S. Cell Signalling by integrins and immunoglobulin Receptor in Primed Meutrophils. trends in Biochem Sci 1995;20(9):362-7.
- Yao m, Sheng ZY, Tian HM, Yu Y, Wang YP, Yang HM. et al. The Association of circulating endotoxemia with the development of multiple organ failure in burned patients. burns 1995;21(4):255-8. o
- Henderson B, Pool S. Modulation of cutokine function: therapeutic applications. En: August V, Anders M, Murad F, eds. Advances in Pharmacology. San Diego: Academic Press, 1994;25:53-96.
- Yentis SM. Cytokines and Sepsis: Time for Reappraisal. Br J Anaesth 1995;74(2):119-20.
- Harris BH, Gelfand JA. The immune response to trauma. Semin Pediatr Surg 1995;4(2):77-82.
- Clancy KD, Lorenz K, Hahn E, Christiansen B, Hofmann C, Gamelli RL. Down regulation of tissue specific tumor necrosis factor alpha in the liver and lung after burn injury and endotoxemia. J Trauma 1997; 42(2):169-76.
- R & D Systems. 1995 Catalog. Cytokines and ralated reagents. Folleto comercial y promocional. R & D Systems Inc. Minneapolis. USA, pág 49-50.
- Mileski WJ, rothlien R, Lipsky P. Interference with the function of leukocyte adhesion molecules by monoclonal antibodies: a New approach to burn injury. Eur J Ped Surg 1994;4(4):225-30.
- R & D Systems. 1995 Catalog. Folleto comercial y promocional. Cytokines and related reagents. R & D Systems Inc. Inneapolis. USA, pág. 187-9.
- Yeh FL, Lin WL, Shen HD, Fang RH. Changes in serum tumor necrosis factor alpha in burned patients. Burns 1997;23(1):6-10.
- Liu XS, Yang ZC, Luo ZH, Huang WH, Li A. A Preliminary exploration of the relationship between Tumor Necrosis Factor and monocytic in vitro production of interleukin (IL-1) and internal organ dysfunction in severely burned Patients. Burns 1995;21(1):29-33.
- Liu XS, Luo ZH, Yang ZC, Huang WH, Li An. The Significance of changes in serum tumor necrosis factor (TNF) activity in severely burned Patients. Burns 1994;20(1):40-4.
- R & D Systems. 1996 Catalog. Cytokines and related reagents. Folleto comercial y promocional. R & D Systems Inc. Minneapolis. USA, pág. 14.
- Serotec 1996 Product Guide. Folleto comercial y promocional. Serotec Systems Inc Nmmeapolis. USA pág. 21-2.
- Monden k, Arii S, Ishiguro S. Involvement of ICAM-1 expression on sinusoidal endothelial cell and neutrophil adherence in the reperfusion injury of cold preserved liver. Transplant Proc 1995;27(1):759-61.
- Ahmed NA, Yee J, Giannias B, Kapadia B, Christou NV. Expression of human neutrophil L-Selectin during the systemic Inflammatory Response Syndrome is Partly mediated by Tumor Necrosis Factor Alpha. Arch Surg 1996;131(1):31-5.
- Parke A, Parke DV. The Pathogenesis of the inflammatory disease: surgical shock and multiple systemic organ failure. Inflammopharmacology 1995;3/2:149-68.
- Zhou DH. Inhibitory effect of albumin or crystalloid resuscitation on bacterial translocation and endotoxin absorption following experimental burn injury. J Surg Res 1992;52:161-6.
- Yamada Y, Endo S, Inada K. Plasma cytokine levels in patients with severe burn injury with reference to the relationship between infection and prognosis. Burns 1996;22(8):587-93.
- Kettle A, Winterbourn C. Assays for the chlorination activity of myeloperoxidase. Meth Enzymnol 1994;233:502-12.
- Tanjoh K, Shima A, Aida M, Tomita R, Kurosu Y. Nitric oxide and active oxygen species in severe sepsis and surgically stressed patients. Surg Today 1995;25(9):774-7.
- Parrillo J. Septic shock in humans. Ann Intern Med 1990;113(3):227-42.
Recibido: 29 de septiembre de 1998. Aprobado: 19 de julio de 1999.
Dr. Félix Broche Valle. Instituto de Ciencias Básicas y Preclínicas "Victoria de Girón". Calle 146 No. 3102, Playa, Ciudad de La Habana, Cuba

