










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Investigaciones Biomédicas
versión On-line ISSN 1561-3011
Rev Cubana Invest Bioméd v.18 n.3 Ciudad de la Habana sep.-dic. 1999
Trabajos de Revisión
Instituto Superior de Ciencias Médicas de CamagüeyMetabolismo de la homocisteína y su relación con la aterosclerosis
Dr. Arturo Menéndez Cabezas y José E. Fernández-Britto RodríguezRESUMEN
Se realizó una revisión sobre los detalles del metabolismo de la homocisteína, aminoácido azufrado que se forma normalmente a partir de la metionina durante el cumplimiento de su función de donante de grupos metilos. Se analizaron sus posibles destinos metabólicos, en particular la remetilación y la transulfuración, en las que están implicadas las formas coenzimáticas de las vitaminas folacina, B12 y B6; así como su oxidación con lo que se origina la homocistina y disulfuros mixtos que incluyen a la llamada homocisteína ligada a proteína, forma principal que circula en el plasma, y otros destinos descritos en la literatura. Se relacionan los métodos de estimación de su concentración plasmática y sus valores de referencia. Se analizaron las posibles causas de hiperhomocisteinemia y los mecanismos fisiopatológicos que vinculan a este estado con la aterogénesis y que tratan de explicar la relación hiperhomocisteinemia-aterosclerosis, propuesta fundamentada por los resultados de estudios epidemiológicos y clínicos de los últimos 30 años.Descriptores DeCS: HOMOCISTEINA/ metabolismo; ATEROSCLEROSIS.
En los últimos años aparecen cada vez con más frecuencia en la literatura médica mundial reportes, artículos originales y revisiones acerca de la relación entre la hiperhomocisteinemia y la aterogénesis. Por lo tanto, es de interés realizar una revisión sobre el metabolismo de este aminoácido, los factores o circunstancias que pueden provocar o evolucionar con elevación de su concentración plasmática y los posibles mecanismos que lo relacionan con el complejo fenómeno de la aterosclerosis.
La homocisteína es un aminoácido azufrado originado metabólicamente de la metionina, aminoácido esencial que, aparte de ser precursor y componente de péptidos y proteínas, desempeña una importante función metabólica al participar en un sistema de transferencia de grupos metilos. En la figura 1 se representa la secuencia de reacciones de este sistema. La metionina, luego de ser activada, sede su grupo metilo en una reacción catalizada por una metiltransferasa, y da lugar a la S-adenosilhomocisteína, la cual se desembaraza por hidrólisis de la adenosina, y se obtiene homocisteína libre.
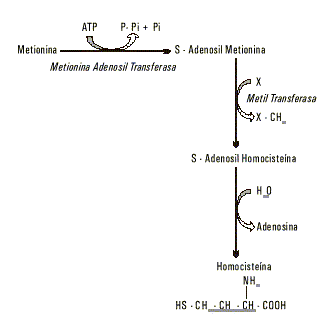
Fig. 1. Formación de homocisteína a partir de metionina.
Destinos de la homocisteína
La homocisteína es metabolizada fundamentalmente a través de 2 posibles vías: la remetilación y la transulfuración.1,2,3La vía de remetilación permite la recuperación de metionina (fig.2). Se trata de una reacción catalizada por la homocisteína metiltransferasa (también denominada metionina sintasa) y representa un punto metabólico con peculiaridades que le confieren singular importancia:
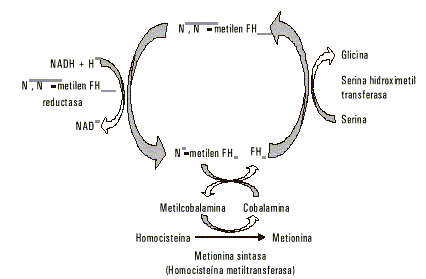
Fig. 2. Recuperación de metionina por la vía de remetilación.
- Posee características de ciclo metabólico con la participación de varios cofactores y enzimas.
- Se produce una interesante interrelación entre cofactores derivados de vitaminas del complejo B, la vitamina B12, que en forma de metilcobalamina es el donante directo del qrupo metilo a la homo-cisteína; la folacina, que en forma de N5-metiltetrahidrofolato sirve de fuente del grupo metilo para la formación de la metilcobalamina; y la vitamina B6, en la forma de fosfato de piridoxal (PLP), como cofactor en el proceso de regeneración del N5-metiltetrahidrofolato.
Otros posibles destinos de la homocisteína
El grupo tiol le confiere a este metabolito la posibilidad de múltiples interacciones y por tanto diversos destinos (fig. 3). Por oxidación 2 moléculas de homocisteína se condensan mediante la formación de un puente disulfuro, y se obtiene así la homocistina.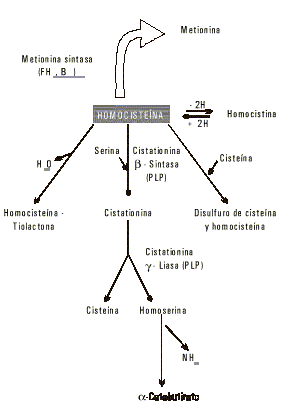
La formación de puentes disulfuro puede ocurrir con otros metabolitos que posean grupo tiol, esto pasa fundamentalmente con la cisteína, y se obtienen disulfuros mixtos de homocisteína con cisteína libre o con restos de cisteína de péptidos y proteínas. A esta última variante se le conoce como homocisteína ligada a proteína,1,2 la forma predominante de la homocisteína circulante.4 Otra posibilidad es que por pérdida de una molécula de agua se obtenga la tiolactona de homocisteína.2
En muchos de los artículos consultados se utilizan los términos homocist(e)ina plasmática y homocist(e)inemia, se entiende por tal al conjunto o concentración en plasma de homocisteína libre, homocistina, disulfuros mixtos con cisteína, homocis-teína ligada a proteína y tiolactona de homocisteína.2,5,6 Coincidimos con Jacobsen1 en que esta forma de designar a las múltiples variantes de la homocisteína circulante es arcaica e incorrecta desde el punto de vista gramatical e incluso práctico, por tanto lo correcto sería utilizar el término homo-cisteína total plasmática u homocisteinemia.
Como se verá más adelante, en la mayoría de los estudios clínicos relacionados con este metabolito se determina la concentración plasmática de homocisteína total, o sea, del referido pool, del cual casi 70 a 90 % corresponde a la ligada a proteína, de 5 a 10 % a la homocistina, de 5 a 10 % a homocisteína-cisteína, y sólo alrededor de 1 % a la homocisteína reducida.1,2 La no ligada a proteína es filtrada en los glomérulos renales, la mayor parte es reabsorbida en los túbulos renales, por lo que sólo muy pequeñas cantidades se excretan por la orina.7
La homocisteína es por tanto un producto normal del metabolismo de la metionina que no circula en grandes cantidades, pues puede ser reciclada a través de la vía de recuperación de la metionina o de la vía de formación de cisteína. Este metabolito en la circulación general y en los tejidos, por su grupo tiol tiende a formar puentes disulfuro, tanto entre sus moléculas como con las de otros compuestos de grupo -SH. De modo que puede considerarse a la homocisteína y los disulfuros que ella forma como pares redox: forma reducida-homocisteína, formas oxidadas-disulfuros (homocistina, homocisteína-cisteína).
Aparte de estos destinos metabólicos descritos, resulta de interés una vía específica en células endoteliales que implica al óxido nítrico.8 Este interesante mensajero químico en presencia de oxígeno reacciona con el grupo sulfhidrilo de la homocisteína y forma la S-nitroso homo-cisteína, y así se bloquean las posibles reacciones de ese grupo (fig. 4).
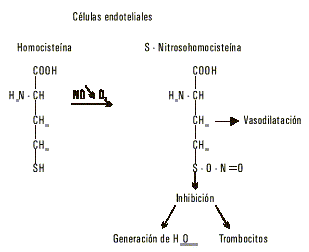
Fig.4. Reacción del óxido nítrico (NO) en presencia de oxígeno
Determinación de la homocisteína plasmática y valores de referencia
Los métodos de estimación de los niveles plasmáticos o séricos de homocisteína total comienzan a desarrollarse a mediados de la década de los años 80 mediante técnicas relativamente complejas y costosas que en general consisten en:1,9-15- Generar homocisteína libre por reducción de los puentes disulfuros mediante la utilización de diferentes agentes reductores.
- Separar la homocisteína de otros metabolitos de bajo peso molecular con grupo tiol mediante cromatografía líquida de alta resolución o por cromatografía gaseosa capilar.
- Determinar la homocisteína mediante detección electroquímica, espectro-fotometría de masa o la fluorimetría previo marcaje con un fluorocromóforo.
- Utilizar anticuerpos monoclonales por inmunoensayo.
En un reporte reciente del Instituto de Nutrición y Tecnología de los Alimentos de Chile, en 128 adultos (22 a 78 años) supuestamente sanos, los valores plasmáticos encontrados fueron de 9,7 ± 6,0 µmol/L para los hombres y de 7,0 ± 3,1 ?mol/L para las mujeres.16
Las personas con valores en ayunas por encima de 15 µmol/L (el percentil 95) son consideradas como portadoras de hiperhomocisteinemia,3,6,17 la cual, según Kang y otros puede ser clasificada en moderada (15 a 30 µmol/L), intermedia (31 a 100 µmol/L y severa (>100 µmol/L ).17 No obstante, Jacobsen1 plantea que se hablaría de "valores deseables" que serían menores que 10 µmol/L.
En los casos de pacientes en los que se sospecha la posibilidad de algún trastorno relacionado con este aminoácido y su nivel plasmático en ayunas se encuentra dentro del rango "normal", se ha utilizado la prueba de sobrecarga de metionina. En este caso, luego de la ayuna nocturna se mide la concentración plasmática basal y pasadas 4 a 8 h del consumo de una solución de metionina (100 mg por kg de peso corporal). Se considera que el paciente es portador de hiperhomocisteinemia, si el valor plasmático luego de la sobrecarga sobrepasa las 2 desviaciones estándar (2DE) del valor de la media de las estimaciones en ayunas.18
Patogenia de la hiperhomocisteinemia
En general se acepta que los determinantes de la hiperhomocisteinemia son complejos e incluyen factores muy diversos de carácter demográfico, genético, adquiridos y que tienen que ver con el estilo de vida.1 En un intento de clasificación patogénica pueden plantearse 3 grupos de causas:- De origen genético.
- Por deficiencias nutricionales.
- Otras causas.
Hiperhomocisteinemias de origen genético
Homocistinuria congénita (homo-cistinuria I o clásica): Se trata de un error congénito del metabolismo con patrón autosómico recesivo, bastante raro (1:200 000 nacimientos), en el cual hay una deficiencia de la cistationina b -sintasa, la principal enzima de la vía de transul-furación. Se caracteriza por una hiperhomo-cisteinemia severa de hasta 500 m mol/L en ayunas, con homocistinuria y varias manifestaciones clínicas, entre las que se destacan la aterosclerosis prematura con pronóstico negativo.19,20 Los pacientes heterocigóticos desarrollan sólo una hiperhomocisteinemia entre moderada e intermedia.20,21Deficiencia de la N5, N10-metileno tetrahidrofolato reductasa (homocistinuria II): Los pacientes homocigóticos con deficiencia de esta enzima de la vía de remetilación desarrollan también una hiperhomocisteinemia severa, con un pronóstico incierto por causa en parte de la ausencia total de una terapéutica efectiva.22,23
Variante termolábil de la N5, N10 -met-leno tetrahidrofolato reductasa: En 1988 Kang y otros24,25 reportaron una variante de la enzima N5, N10-metileno tetrahidrofolato reductasa (D -MTHFR) con actividad disminuida, causada por una mutación puntual (677 C T) que produce una sustitución de un resto de valina por uno de alanina. Esta mutación ha sido encontrada en 38 % de los francoca-nadienses, en 5 a 15 % de la población general de Canadá,2 en 25 a 39 % de otros grupos poblacionales1,2,5,26 y en sólo 10 % de los afronorteamericanos.27 Los homocigóticos presentan hiperhomocisteinemia moderada o intermedia y se ha planteado su posible relación con los defectos del tubo neural.28 Resulta interesante el hallazgo reciente de otra variante de la enzima por una mutación 1298 A C, resultante en la sustitución de un resto de glutamato por uno de alanina y una disminución de la actividad catalítica, pero no se acompaña de hiperhomocisteinemia. Sin embargo, en los heterocigóticos combinados en ambas mutaciones se observa una marcada reducción de la actividad de la enzima e hiperhomocisteinemia, y se reporta en 28 % de los recién nacidos estudiados con defectos del tubo neural.29 Estos y otros posibles casos de mutaciones del gen de la enzima MTHFR pueden presentar una interesante combinación de factores patogénicos de carácter genético y ambiental, ya que si al debilitamiento de la actividad de la enzima se añade el déficit de la folacina, es lógico que se desarrolle una hiperhomocisteinemia con sus probables consecuencias.30,31
Errores innatos del metabolismo de la vitamina B12: Se trata de alteraciones de origen genético de la absorción, el transporte y la activación de la cobalamina, que traen como consecuencia una reducción de la actividad de la metionina sintasa y por ende hiperhomocisteinemia.32 Aunque también se han descrito mutaciones del gen de la metionina sintasa, aún queda por aclarar su prevalencia y su contribución a la elevación plasmática de homocisteína en pacientes heterocigóticos.1
Hiperhomocisteinemias por deficiencias nutricionales
Hasta el presente se conoce que la deficiencia individual o combinada de 3 vitaminas del complejo B, la B12, la folacina y la B6 puede ser causa de hiperhomocisteinemia, lo cual es lógico al considerar la participación de los cofactores derivados de estas vitaminas en las 2 principales vías metabólicas de la homocisteína.Existen numerosos reportes de elevada concentración plasmática de homocisteína total en pacientes con deficiencia nutricional de cobalamina y folacina, así como de la correlación negativa entre los niveles séricos de folato, vitaminas B12 y B6 y los valores plasmáticos de homocisteína.33-35 Algnos autores han llegado a sugerir que los niveles séricos bajos de 1 o de las 3 vitaminas son factores coadyuvantes en casi dos terceras partes de todos los casos de hiperhomocisteinemia.34
Por otra parte, la suplementación con una, fundamentalmente folacina, o diferentes combinaciones de estas vitaminas ha resultado exitosa en la reducción de los niveles plasmáticos de homocisteína total.36,37 Si tenemos en cuenta los principales alimentos que sirven de fuente de la folacina y los hábitos alimentarios prevalentes en las sociedades occidentales, resulta evidente que predomina la paradoja de bajo consumo de esa vitamina y el predominio de alimentos relativamente ricos en metionina; con esto se propicia la elevación de homocisteína circulante en el período posprandial, y, por tanto, la posibilidad de deterioro de la función endotelial de los vasos sanguíneos.1,38
Otras causas de hiperhomocisteinemia
Enfermedad renal crónica: Los niveles plasmáticos de homocisteína se elevan de 2 a 4 veces en pacientes con insuficiencia renal crónica, lo cual se correlaciona con la concentración sérica de creatinina y albúmina.7 No obstante, aún no está claro si la hiperhomocisteinemia de los pacientes renales en estadio final se debe a una reducción de la excreción o a la alteración de la metabolización del aminoácido en las células renales.2Hipotiroidismo: Se ha reportado la presencia de hiperhomocisteinemia en pacientes hipotiroideos sin que haya una explicación clara del porqué.1,2 En un reporte reciente39 los autores encontraron valores mayores de homocisteína plasmática en pacientes hipotiroideos en comparación con pacientes hipertiroideos y con los individuos de un grupo control. Resulta interesante que los pacientes con hipofunción tiroidea también presentaron niveles superiores de creatinina sérica que los controles.
Anemia perniciosa: Teniendo en cuenta el papel de la cobalamina en la vía de remetilación, es lógico que en los pacientes con insuficiente absorción de esa vitamina se produzca elevación plasmática de homocisteína.2
Cáncer: Se han reportado altos niveles plasmáticos de homocisteína en pacientes con diferentes tipos de carcinoma, fundamentalmente de mama, ovario y páncreas; así como en casos de leucemia linfoblástica aguda. Esto puede explicarse a partir de la observación experimental de que células transformadas en cultivo no son capaces de metabolizar la homocisteína.2,40,41
Drogas o fármacos: En el arsenal terapéutico actual se cuenta con algunas drogas que de una u otra forma pueden causar elevación de la concentración plasmática de homocisteína. El metotrexate, droga anticancerosa, tiene potente acción inhibitoria de la folato reductasa, enzima clave en la activación de la folacina, de ahí que el consumo de este fármaco provoque aumentos no permanentes de la homocisteína circulante.42 La fenitoína (difenilhidantoína), anticonvulsivante de acción en el nivel de la corteza motora, aparte de su acción principal se ha establecido su interferencia con el metabolismo del folato, lo que explica la hiperhomocis-teinemia moderada observada durante su uso terapéutico.2,42La teofilina, de la familia conocida de las metilxantinas, cuya acción inhibitoria de la fosfodiesterasa es bien conocida, puede causar hiperhomocisteinemia por interferir en la síntesis de piridoxal fosfato, la forma coenzimática de la vitamina B6.2 Se ha sugerido que el colestipol y el ácido nicotínico, drogas reductoras del nivel circulante de lípidos, pueden elevar los niveles plasmáticos de homocisteína por alteración de la absorción de folato.43
Hábitos tóxicos: En el estudio Hordaland en Noruega se encontró asociación entre el tabaquismo y excesivo consumo de café y la aparición de hiperhomocisteinemia, probablemente por interferencia con la síntesis de piridoxal fosfato.44,45 También el alcoholismo crónico se ha reportado como causal de elevación plasmática de homocisteína, esto parece deberse a los déficits nutricionales de esos pacientes.46
Pacientes con transplante cardíaco: Los receptores de transplante de corazón presentan hiperhomocisteinemia moderada a intermedia, lo que en parte puede estar relacionado con insuficiencia renal.47
Relación de la hiperhomocisteinemia con la aterosclerosis
Aunque ya en 1968 Carey y otros,20 al describir el cuadro clínico de 9 pacientes homocigóticos con homocistinuria I, habían destacado las complicaciones aterotrombóticas de aparición temprana, no es hasta el año siguiente que KS McCully48 plantea la hipótesis de que la hiperhomocisteinemia puede tener relación causal con la aterosclerosis.En los casi 3 decenios transcurridos desde entonces han sido numerosos los estudios que han tratado de esclarecer esta posible asociación, sobre todo a partir del desarrollo de métodos cada vez más sensibles y específicos para la estimación de homocisteína. Welch y Loscalzo2 afirman que más de 20 estudios casos control y longitudinales han validado esta relación.
Por su parte Kronenberg,7 investigador estudioso de los factores metabólicos relacionados con las complicaciones cardiovasculares de los pacientes con enfermedad renal crónica, hace referencia a 3 estudios prospectivos recientes que demuestran una asociación fuerte entre altas concentraciones plasmáticas de homocisteína y alteraciones ateroscleróticas.
Por lo tanto, más de 80 estudios de carácter epidemiológico con enfoque de riesgo en los últimos 10 años1 confirman esta relación; lo que aún queda por aclarar es si realmente la homocisteína, o más bien, la hiperhomocisteinemia, es un factor causal o un elemento acompañante de la aterosclerosis, precisar cuáles son los mecanismos patofisiológicos subyacentes, y por último, si su corrección puede servir de prevención o de tratamiento eficaz de sus graves consecuencias, como son la enfermedad coronaria, los accidentes vasculares encefálicos y los eventos vasculares periféricos.
Mecanismos patofisiológicos probables
Numerosos estudios con modelos in vitro, así como con animales de experimentación in vivo, y algunas investigaciones clínicas han tratado de establecer los mecanismos íntimos de la posible relación causaefecto entre hiperhomocisteinemia y aterogénesis. Es necesario aclarar que la inmensa mayoría de los estudios in vitro, fundamentalmente con cultivos de células endoteliales y musculares lisas de vasos sanguíneos obtenidos de animales y humanos, utilizan concentraciones de homocisteína muy altas (5-10 nmol/L), muy por encima de los valores fisiológicos y patofisiológicos, además, en muchos falta la confirmación de la especificidad para la homocisteína de los cambios observados, y queda la duda de si el efecto se debe al grupo tiol de cualquier molécula que lo posea;1 de ahí que toda esta información deba interpretarse con la necesaria cautela y crítica. A partir del análisis de la literatura revisada se ha intentado resumir y sistematizar el conjunto de evidencias y hallazgos, los cuales se relacionan con los aspectos etiológicos y fisiopatológicos más aceptados del complejo fenómeno aterosclerótico, o sea, en correspondencia con la hipótesis de Ross de "la respuesta a la lesión"49 que implica a las células endoteliales, las células musculares lisas, las células blancas, los trombocitos y un conjunto de biomoléculas que participan en un intrincado proceso crónico cada vez mejor caracterizado. Aunque se examinan los posibles mecanismos por separado se hará evidente que más bien se trata de un conjunto de vías y efectos concatenados.Daño oxidativo
Durante la autoxidación de homocisteína se generan potentes especies reactivas del oxígeno, como son el anión superóxido, el peróxido de hidrógeno y el anión hidroxilo,1,2,8,50,5 los que a su vez pueden provocar difunción endotelial, con el consiguiente daño de la pared vascular y sus graves consecuencias (regulación vasomotora alterada, cambio del fenotipo antitrombótico, activación y agregación plaquetaria, activación de la elastasa, aumento de la deposición de Ca2+ en la íntima arterial) y peroxidación de los lípidos de las lipoproteínas plasmáticas, fundamentalmente de las lipoproteínas de baja densidad (LDL), con la formación de hidroxicolesteroles altamente aterogénicos, la degradación de ácidos grasos polinsaturados, la formación de lisolecitina y la modificación aldehídica de los restos de lisina de la Apo B100, con los consiguientes efectos citotóxicos y aterogénicos.51,52Efectos directos de la homocisteína y de la homocistina
Se ha reportado el efecto estimulante de la homocisteína sobre la síntesis de ADN en células musculares lisas (CMLs) en cultivo, obtenidas de vasos sanguíneos de humanos.53 El efecto es bifásico, o sea, primero se observa un incremento de la intensidad de la biosíntesis de ADN, en la medida en que aumenta la concentración de homocisteína en el medio de cultivo de CMLs, seguido de inhibición de este proceso a concentraciones mucho mayores, que sobrepasan los límites patofi-siológicos.Un efecto bifásico de la homocisteína se ha observado también sobre la actividad colagenasa medida por zimografía en la matriz extracelular de la pared vascular.54
También se reporta la inhibición o el bloqueo que ejerce la forma reducida de este aminoácido sobre la interacción del activador tisular de plasminógeno con la anexina II,55 esto puede provocar cambio del patrón antitrombótico de las células endoteliales de la pared vascular.
En su reciente revisión Jacobsen1 especula sobre un hallazgo de su grupo: células endoteliales aórticas de humanos en cultivo no expresan la forma activa de la cistationina b -sintasa (CBS), por lo que estas células no pueden iniciar el catabolismo de la homocisteína por la vía de la transulfuración, haciéndolas más susceptibles a los incrementos de su concentración.
El mismo autor reporta que su grupo de investigación demostró que concentraciones de homocisteína del orden de los 10 a 50 m mol/L estimulan la expresión de la proteína quimiotáctica-1 (PQ-1) en células endoteliales aórticas en cultivo, efecto que no se logró con otros amino-ácidos azufrados (cisteína, cistina, metionina y homocistina).1 La PQ-1 es un potente agente quimiotáctico para monocitos de la familia de las Quimiocinas C-C, por lo que este resultado apunta a uno de los fenómenos fisiopatológicos de la aterosclerosis, la respuesta inflamatoria de la pared vascular, en este caso producida por la elevada concentración de homocisteína.
Por otro lado, resultan de interés los resultados experimentales de Tyagi,56 quien trató de demostrar que la forma oxidada de la homocisteína, la homocistina, puede causar alteraciones ateroscleróticas prematuras en el corazón. El autor cultivó células musculares lisas aisladas de las arterias coronarias de corazones humanos con cardiomiopatía idiopática. La adición de homocistina a los cultivos produjo un evidente aumento del número de CMLs, o sea, se estimuló la proliferación de estas células, lo cual fue bloqueado por la adición del antioxidante N-acetil cisteína (NAC). También la homocistina indujo la síntesis de colágeno en una forma dependiente del tiempo y de la dosis, efecto que se logró inhibir al añadir al medio de cultivo NAC o glutatión reducido. Y, por último, logró demostrar capacidad de ligar homocistina por proteínas de 25 a 40 kDa encontradas en la membrana plasmática y el citosol de estas células en cultivo, efecto que fue inhibido por la NAC. El autor sugiere que estas proteínas pueden ser receptores específicos para la homocisteína oxidada, con esto se abre un interesante campo de investigacón.
Efectos sobre el metabolismo del óxido Nítrico (no)
La exposición por largo tiempo de las células endoteliales a la homocisteína puede producir una disminución de la disponibilidad de NO por 2 vías: por afectación de su síntesis,50,57 directamente o mediado por especies reactivas del oxígeno o productos de la peroxidación lipídica, y por agotamiento del gas ya formado al aumentar la posibilidad de formar S-nitrosoho-mocisteína,8 con esto se crea una especie de círculo vicioso que agrava aún más el daño oxidativo y bloquea el efecto vasodilatador de este peculiar mensajero químico.Sin embargo, en las células musculares lisas es otro el efecto, Welch y otros58 reportan que la homocisteína promueve directamente o mediada por especies reactivas del oxígeno, el aumento de la producción de NO en estas células por inducción de la sintetasa de óxido nítrico 2 (NOS 2) medida por el NFkB, lo cual habla a favor de la acción mitogénica de la homocisteína, pues se ha demostrado el papel del mencionado factor de transcripción para la proliferación de células musculares lisas en cultivo.59
Alteraciones dependientes de la tiolactona de homocisteína
La tiolactona de homocisteína casi siempre se produce en ínfimas cantidades, pero al aumentar significativamente la concentración del aminoácido circulante puede incrementarse su formación y puede conducir a las situaciones siguientes:- Combinación con las partículas de LDL, lo que trae como consecuencia su agregación y captación por los macrófagos de la ínfima arterial y las células espumosas de las placas de ateroma en formación.2,33
- Conjugación con proteínas intrace-lulares y de secreción, a través de la acilación de restos de lisina por el carboxilo activado de la tiolactona, lo que conduce a alteraciones del metabolismo oxidativo, con esto se refuerza el daño oxidativo, y cambios fibróticos y proliferativos de las CMLs de la pared vascular.60
No hay dudas de que a pesar de la enorme relación de hallazgos experimentales, es aún arriesgado aseverar que hay una relación causaefecto indiscutible entre hiperhomocisteinemia y aterosclerosis, pero sí es incuestionable que esta condición puede ser considerada como un factor de riesgo más de este complejo proceso patológico inherente a la vida del hombre. El conocimiento de las causas y factores que favorecen la elevación de este metabolito y sus derivados en la circulación general y los tejidos, así como las posibles vías de su corrección, en lo fundamental a través de la suplementación vitamínica y de correctos hábitos dietéticos y, en definitiva de un estilo de vida sano -aunque no se haya probado científicamente que la disminución de la concentración plasmática de la homocisteína total logre detener los procesos ateroscleróticos y sus principales manifestaciones- introduce nuevos retos a las Ciencias Médicas, al enfoque de este flagelo y sobre todo a los aspectos de prevención y promoción de salud.
SUMMARY
A review on the details of the metabolism of homocysteine, a sulfur-containing amino acid that is normally obtained from methionine during the accomplishment of its function as a donor of methyl groups was made. Its possible metabolic destinations are analyzed, particularly the remethylation and the transsulfuration, in which the coenzymatic forms of vitamins B6, B12 and folate are involved, as well as its oxidation with which homocystine and mixed disulphides including the so-called homocystein attached to protcin, which is the main form circulating in plasma and in other destinations described in literature, are originated. The methods for stimating its plasmatic concentration and its reference values are related. The possible cause of hyperhomocysteinemia and the physiopathological mechanisms that link this status to atherogenesis and that try to explain the proposed hyperhomocysteinemia- atherosclerosis relation established by the results of epidemiological and clinical studies during the last 30 years are analyzed.Subject headings: HOMOCYSTEINE/metabolism; ATHEROSCLEROSIS.
REFERENCIAS BIBLIOGRÁFICAS
- Jaconsen DW. Homocysteine and vitamins in cardiovascular disease. Clin Chem 1998; 44: 1833-43.
- Welch GN, Loscalzo J. Homocysteine and atherothrombosis. N Engl J Med 1998;338;1042-50.
- Miner SE, Evroski J, Cole DE. Clinical chemistry and molecular biology of homocysteine metabolism: an update. Clin Biochem 1997;30:189-201.
- Ueland PM. Homocysteine species as components of plasma redox thiol status. Clin Chem 1995;41:340-2.
- Malinow MR. Plasma homocysteine: a risk factor for arterial occlusive diseases. J Nutr 1996;126:1238S- 43S.
- Superko HR. New aspects of risk factors for the development of atherosclerosis, including small low-density lipoprotein, homocyst (e) ine, and lipoprotein (a). Curr Opin Cardiol 1995;10:347-54.
- Kronenberg F. Homocysteine lipoprotein (a) and fibrinogen: metabolic risk factors for cardiovascular complications of chronic renal disease. Curr Opin Nephrol Hypertens 1998;7:271-8.
- Stamler JS, Osborne JA, Jaraki O, Simon DI, Welch GN, Upchurch GR Jr, Loscalzo J. Adverse vascular effects of homocysteine are modulated by endothelium-derived relaxing factor and related oxides of nitrogen. J Clin Invest 1993;9:308-18.
- Ueland PM, Refsum H, Stabler SP, Malinow MR, Andersson A, Allen RH. Total homocysteine in plasma or serum: methods and clinical applications. Clin Chem 1993;39:1764-79.
- Stabler SP, Marcell PD, Podell ER, Allem RH. Quantitation of total homocysteine, total cysteine, and methionine in normal, serum and urine using capillary gas chromatography-mass spectrometry. Anal Biochem 1987;162:185-96.
- Araki A, Sako Y. Determination of free and total homocysteine in human plasma by high-performance liquid chromatography with fluorescence detection. J Chromatogr 1987;422:43-52.
- Andersson A, Isaksson A, Brattström L, Hultberg B. Homocysteine and other thiols determined in plasma by HPLC and thiol-specific postcolumn derivatization. Clin Chen 1993;39:1590-7.
- Fiskerstrand T, Refsum H, Kvalheim G, Ueland PM. Homocysteine and other thiols in plasma and urine automated determination and sample stability. Clin Chem 1993;39:263-71.
- Frantzen F, Faaren AL, Alfheim I, Nordhei AK. Enzyme conversion immunoassay for determining total homocysteine in plasma or serum. Clin Chem 1998;44:311-6.
- Shipchandler MT, Moore EG. Rapid, fully automated measurement of plasma homocysteine with the Abbot Imx analyzer. Clin Chem 1995;41:991-4.
- Bunout D, Petermann M, Maza P, de la, Kauffmann R, Suazo, M, Hirsch S. Niveles séricos de homocisteína en adultos chilenos sanos. Rev Med Chil 1998;126:905-10.
- Kang SS, Wong PW, Malinow MR. Hyperhomocysteinemia as a risk factor for occlusive vascular disease. Ann Rev Nutr 1992;12:279-98.
- Dudman NP, Wilcken DE, Wang J, Lynch JF, Macey D, Lundberg P. Disordered methionine/homocysteine metabolism in premature vascular disease: its occurrence, cofactor therapy, and enzymology. Arterioscl Thromb 1993;13:1253-60.
- Mudd SH, Levy HL, Skovby F. Disorders of transsulfuration. En: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic and molecular basis of inherited disease 7th ed. New York: McGraw-Hill, 1995; vol 1:1279-1327.
- Carey MC, Donovan DE, Fitzgerald O, McAuley FD. Homocystinuria I. A clinical and pathological study of nine subjects in six families. Am J Med 1968;45:7-25.
- Malinow MR, Sexton G, Averbuch M, Wilson O, Upson B. Homocysteine in daily practice: levels in coronary heart disease. Coronary Artery Dis 1990;2:4-12.
- Mudd SH, Uhlendorf BW, Freeman JM, Finfelstein JD, Shih VE. Homocystinuria associated with decreased methylene-tetra hydrofolate reductase activity. Biochem Biophys Res Commun 1972;46:905-12.
- Erbe RW. Inborn errors of folate metabolism. En: Blakely RL, Whitehead VM, eds.Folate and Pterins: nutritional, pharmacological, and physiological aspects. New York: Marcel Dekker,1986:413-25.
- Kang SS, Zhou J, Wong PWK, Kowalisyn J, Strokosch G. Intermediate homocystinuria: a thermolabile variant of methylene tetrahydrofolate reductase. Am J Hum Genet 1988;43:414-21.
- Izumi M, Iwai N, Ohmichi N, Nakamura Y, Shimoike H, Kinoshita M. Molecular variant of 5,10- methylene tetrahydrofolate reductase: risk factor of ischemic heart disease in the Japanese population. Atherosclerosis 1996;121:293-4.
- Wilcken DEL, Wang XL, Sim AS, McCredie RM. Distribution in healthy and coronary populations of the methylene tetrahydrofolate reductase (MTHFR) C677T mutation. Arterioscler Thromb Vasc Biol 1996;16:878-82.
- McAndrew PE, Brandt JT, Pearl DK, Prior TW. The incidence of the gene for themolabile methylene tetrahydrofolate reductase in African Americans. Thromb Res 1996;83:195-8.
- Wagner WE, Levine B. Folic acid and neural tube defects. Curr Concepts Nutr 1993;8:1-12.
- Put NM, Van der, Gabreels F, Stevens EM, Smeitink JA, Trijbels FJ, Eskes TK, et al. A second common mutation in the methylene tetrahydrofolate reductase gen: an additional risk factor to neural tube deffects? Am J Hum Genet 1998;62:1044-51.
- Girelli D, Friso S, Tiabetti E, Olivieri O, Russo C, Pessotto R, et al. Methylene tetrahydrofolate reductase C677 T mutation, plasma homocysteine, and folate in subjects from northen Italy with or without angiographically documented severe coronary atherosclerotic disease: evidence for an important genetic-environmental interaction. Blood 1998;91:4158-63.
- Zittoun J, Tonetti C, Bories D, Pignon JM, Tulliez M. Plasma homocysteine levels related to interactions between folate status and methylene tetrahidrofolate reductase: a study in 53 healthy subjects. Metabolism 1998;47:1413-8.
- Fenton WA, Rosenberg LE. Inherited disorders of cobalamin transport and metabolism. En: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic and molecular basis of inherited disease. 7th ed. New York: McGraw-Hill, 1995:3129- 49.
- Verhoef P, Stampfer MJ, Buring JE, Gaziano JM, Allen RH. Stabler SP, et al. Homocysteine metabolism and risk of myocardial infarction: relation with vitamins B6 , B12, and folate. Am J Epidemiol 1996;143:845-59.
- Selhub J, Jacques PF, Wilson PWF, Rush D, Rosenberg IH. Vitamin status and intake as primary determinants of homocysteinemia in an elderly population. JAMA 1993;270:2693-8.
- Ubbink JB, Vermack WJH, Merwe A. van der, Becker, PJ. Vitamina B12, Vitamin B6, folate nutritional status in men with hyperhomocysteinemia. Am J Clin Nutr 1993;57:47-53.
- Den Heijer M, Brouwer IA, Bos GM, Blom HJ, Put NM van der, Spaans AP, et al . Vitamin supplementation reduces blood homocysteine levels: a controled trial in patients with venous thrombosis and healthy volunteers. Arterioscler Thromb Vasc Biol 1998;18:356-61.
- Hirose S, Kim S, Matsuda A, Itakura Y, Matsumura O, Tamura H, et al . Effects of folic acid supplementation on hyperhomocysteinemia in CAPD patients: effects on unsaturated fatty acids. Nippon Jinzo Gakkai Shi 1998;40:8-16.
- Bellamy MF, McDowell IF, Ramsey MW, Brownlee M, Bones C, Newcombe RG, et al. Hyperhomocysteinemia after an oral methionine load acutely impairs endothelial function in healthy adults. Circulation 1998;98:1848-52.
- Nedrebo BG, Ericsson UB, Nygard O, Refsum H, Ueland PM, Aakvaag A, et al. Plasma total homocysteine levels in hyperthyroid and hypothyroid patientes. Metabolism 1998;47:89-93.
- Mayer EL, Jacobsen DW, Robinson K. Homocysteine and coronary atherosclerosis. J Am Coll Cardiol 1996;27:517-27.
- Refsum H, Wesenberg F, Ueland PM. Plasma homocysteine in children with acute lymphoblastic leukemia: changes during a chemotherapeutic regimen including methotrexate. Cáncer Res 1991;51:828-35.
- Ueland PM, Refsum H, Brattstrom L. Plasma homocysteine and cardiovascular disease. En: Francis RB Jr, ed. Atherosclerotic cardiovascular disease, hemostasis, and endothelial function. New York: Marcel Dekker,1992:183-236.
- Blankenhorn DH, Malinow MR, Mack WJ. Colestipol plus Niacin therapy elevates plasma homocysteine levels. Coronary Artery Dis 1991;2:357-60.
- Nygard O, Vollset SE, Refsum H, Stensvold I, T verdal A, Nordrehaug JE, et al . Total plasma homocysteine and cardiovascular risk profile- the Hordaland homocysteine study. JAMA 1995;274:1526-33.
- Nyård O, Refsum H, Ueland PM, Stensvold I, Nordrehaug JE, Kvåle G, et al. Coffee consumption and plasma total homocysteine-the Hordaland homocysteine study. Am J Clin Nutr 1997;65:136-43.
- Cravo ML, Gloria LM, Selhub J, Nadeau MR, Camilo ME, Resende MP, et al. Hyperhomocysteinemia in chronic alcoholism: correlation with folate, vitamin B-12, and vitamin B-6 status. Am J Clin Nutr 1996;63:220-4.
- Cole DE,Ross HJ, Evrovski J, Langman LJ, Miner SE, Daly PA, et al. Correlation between total homocysteine and cyclosporine concentrations in cardiac transplant recipients. Clin Chem 1998;44:2307-12.
- McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol 1969;56:11-28.
- Ross R. The pathogenesis of atherosclerosis- an update. N Eng J Med 1986;314:488-500.
- Welch GN, Upchurch GR Jr. Loscalzo J. Hyperhomocysteinemia and atherothrombosis. Ann NY Acad Sci 1997;811:48-58.
- Loscalzo J. The oxidant stress of hyperhomocystein(e)mia. J Clin Invest 1996;98:5-7.
- Heinecke JW, Rosen H, Suzuki LA, Chait A. The role of sulfur-containing amino acids in superoxide production and modification of low density lipoprotein by arterial smooth muscle cells. J Biol Chem 1987; 262:10098-103.
- Tang L, Mamotte CD, Bockxmeer FM, van, Taylor RR. The effect of homocysteine on DNA synthesis in cultured human vascular smooth muscle. Atherosclerosis 1998;136:169-73.
- Tyagi SC, Smiley LM, Mujumdar VS, Clonts B, Parker JL. Reduction-oxidation (Redox) and vascular tissue level of homocysteine in human coronary atherosclerotic lesions and role in extracelular matrix remodeling and vascular tone. Mol Cell Biochem 1998;181:107-16.
- Hajjar KA, Mauri L, Jacovina AT, Zhong F, Mirza UA, Padovan JC, Chait BT. Tissue plasminogen activator binding to annexin II tail domain. Direct modulation by homocysteine. J Biol Chem 1998;273: 9987-93.
- Tyagi SC. Homocysteine redox receptor and regulation of extracellular matrix components in vascular cells. Am J Physiol 1998;274:C396-C405.
- Upchurch GR Jr, Welch GN, Fabian AJ, Brecher P, Loscalzo J, Pigazzi A. Homocysteine decreases bioavailable nitric oxide by a mechanism involving glutathione peroxidase. J Biol Chem 1997;272:17012-7.
- Welch GN, Upchurch GR Jr, Farivar RS, Pigazzi A, Vu K, Brecher P. Homocysteine-induced nitric oxide production in vascular smooth-muscle cells by NF-kappa B-dependent transcriptional activation of Nos 2. Proc Am Assoc Physicians 1998;110:22-31.
- Bellas RE,Lee JS, Sonenshein GE. Expression of constitutive NF-kappa B-like activity is essential for proliferation of cultured bovine vascular smooth muscle cells. J Clin Invest 1995;96:2521-7.
- Jakubowski H. Metabolism of homocysteine thiolactone in human cell cultures: possible mechanism for pathological consequences of elevated homocysteine levels. J Biol Chem 1997;272:1935-42.
- McCully KS. Homocysteine metabolism in scurvy, growth and arterioclerosis. Nature 1971;231:391-2.
Recibido: 1 de junio de 1999. Aprobado: 15 de junio de 1999.
Dr. Arturo Menéndez Cabeza. Ave. Los Ancianos, Edificio No.13, Apto 21. Reparto Previsora, Camagüey.
Correo electrónico:artemen@finlay.cmw.sld.cu

