










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Investigaciones Biomédicas
versión impresa ISSN 0864-0300
Rev Cubana Invest Bioméd vol.31 no.2 Ciudad de la Habana abr.-jun. 2012
ACTUALIZACIÓN
Actualización sobre el síndrome de QT largo congénito
Update on the congenital long QT syndrome
Dr. Roberto Zayas Molina
Instituto de Cardiología y Cirugía Cardiovascular.
RESUMEN
El síndrome de QT largo congénito se caracteriza por la prolongación del intervalo QT y anomalías de la onda T del electrocardiograma, asociados con una predisposición para la aparición de arritmias ventriculares malignas (torsión de puntas), síncope, fibrilación ventricular y muerte súbita (MS) cardíaca. El entrenamiento para realizar las mediciones, la variabilidad de la penetración y de las mutaciones, los solapamientos, las formas mixtas y los síndromes de superposición, pueden dificultar el diagnóstico y con ello, la conducta terapéutica. Esta revisión pretende actualizar aspectos esenciales relacionados con el diagnóstico, bases genéticas, mecanismos fisiopatológicos y moleculares, así como conducta terapéutica, de esta compleja canalopatía.
Palabras clave: QT largo.
ABSTRACT
The congenital long QT syndrome is characterized by a prolongation of the QT interval and T wave abnormalities on the ECG, associated to a predisposition for the appearance of malignant ventricular arrhythmias (torsade de pointes), syncope, ventricular fibrillation and sudden cardiac death (SCD). Training in taking the measurements, penetrance and mutation variability, overlaps, mixed forms and overlap syndrome may all hamper diagnostic and therapeutic management. The present review intends to update essential aspects related to the diagnosis, genetic bases, physiopathological and molecular mechanisms, and therapeutic management of this complex channelopathy.
Key words: long QT.
INTRODUCIÓN
El síndrome de QT largo (SQTL) es una canalopatía de origen genético que se asocia con prolongación del intervalo QT y a anomalías de la onda T en el electrocardiograma (ECG) de 12 derivaciones, con predisposición para la aparición de arritmias ventriculares polimórficas (torsión de puntas), síncope, fibrilación ventricular, paro cardíaco y muerte súbita (MS) cardíaca.
En 1957 Jervell y Lange-Nielsen,1 (J L-N) describen la primera familia con SQTL congénito, en niños con sordera bilateral, síncopes recurrentes, MS cardíaca y QT largo (QTL); este trastorno tiene un patrón de herencia autosómico recesivo. Romano2 y Ward,3 años más tarde, presentan una forma familiar más común, con un cuadro clínico similar al anterior, sin sordera, que se trasmite con un patrón de herencia autosómico dominante. A partir de entonces, ha aparecido una gran cantidad de publicaciones en diversas revistas internacionales. Priori y otros4 describen en 1998, una variante recesiva del Romano-Ward.
En 1979 se inicia un registro internacional para el SQTL que permite una mejor comprensión y evaluación del curso clínico, de este complejo trastorno de repolarización.5
Esta canalopatía, la primera descrita, tiene un nivel de penetración variable y es una de las principales causas de MS en niños y jóvenes con función cardíaca normal. Aunque se encuentra bien caracterizada, todavía existen limitaciones relacionadas con el diagnóstico, genética y bases moleculares. En este acápite se hace referencia a algunas particularidades relacionadas con las formas hereditarias del SQTL congénito.
PREVALENCIA
En la literatura se encuentran cifras que oscilan entre 1:20006 y 1:20000;7 esta diferencia puede ser el resultado de las limitaciones en el diagnóstico y la incidencia de formas ocultas y mixtas. El J L-N, más raro, tiene una prevalencia de 1:50000.8 Entre el 10 y 12 de los casos de MS infantil pueden ser ocasionados por SQTL.9
Fundamentos y consideraciones del genotipo-fenotipo
El potencial de acción (PA) es el resultado de la compleja y especializada actividad de los canales iónicos que permiten el paso selectivo de las distintas corrientes para garantizar los gradientes a través de la membrana celular y la función contráctil del cardiomiocito. En el SQTL pueden existir disminución de las corrientes de salida de K+ (IKs, IKr, IK1 e IKACh), incremento de las corrientes de entrada Na+ o Ca+2, o pérdida de la función de proteínas regulatorias, de anclaje estructural o caveolin.10,11
Se han descrito más de 700 mutaciones en 13 genes que identifican a 13 tipos de QTL congénitos. A pesar del desarrollo de la genética y de la creciente identificación de nuevos subtipos, más del 75 de las mutaciones se concentran en 3 genes principales:12 KCNQ1 (QTL1), KCNQ2 (QTL2) y SCN5A (QTL3) que se encuentran bien estudiados desde el punto de vista clínico y electrocardiográfico.12-14 El resto de los SQTL son muy raros y los reportes en la literatura casi anecdóticos.
El QTL1 es el subtipo más frecuente, se caracteriza por la pérdida de la función de las mutaciones del KCNQ1 que codifica las IKs; los eventos arrítmicos se relacionan sobre todo con los ejercicios, el estrés físico o emocional; esta relación con el estrés y la descarga adrenérgica determina su respuesta favorable a los β-bloqueadores y la menor necesidad de acudir a terapias más agresivas como el desfibrilador automático (DAI) en los adultos. El ECG de estos casos muestra una T de base ancha.12,14 (Fig. 1).
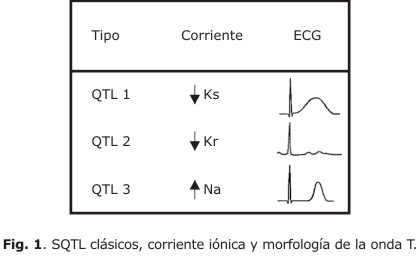
Las mutaciones del KCNH2 (también llamado HERG) son responsables del QTL2 y las segundas en prevalencia, codifican las IKr. Los síntomas se manifiestan con los estímulos auditivos (timbres o alarmas), aunque pueden revelarse también con el estrés emocional y menos frecuentemente con el ejercicio; pueden debutar en el período posparto.12,14 Hasta el 50 o 60 % de las arritmias ventriculares pueden aparecer antes de los 40 años, con una incidencia anual de MS o paro cardíaco de 0,6 %.15 En el ECG se puede encontrar una onda T de poca amplitud que puede estar mellada, con muesca o bifásica.
Las mutaciones del SCN5A (el menos prevalente) origina el QTL3 que se relaciona con el incremento del Na intracelular.14 Los síntomas se asocian con el reposo, sueño o frecuencias cardíacas lentas; la letalidad de este subtipo es mayor, con una elevada incidencia de paro cardíaco o MS y poca respuesta a los b-bloqueadores.12,14 El segmento ST tiene una larga línea isoeléctrica, con una T de base estrecha.12
El gen ANK 2 responsable del LQT4, regula la proteína Ankirin B. Como sus mutaciones se pueden asociar con disfunción ventricular, bradicardia y otras arritmias ventriculares malignas sin que exista QTL, algunos investigadores han sugerido considerar su déficit, como una entidad clínica independiente del SQTL clásico.16
El SQTL congénito ha sido considerado durante décadas una enfermedad eléctrica de origen genético que aparece en el corazón estructuralmente sano; sin embargo, en la literatura aparecen publicaciones que revelan la presencia de trastornos orgánicos asociados.17-19 Recientemente Haugaa y otros,17 demostraron la presencia de anomalías segmentarias en la duración de la contracción ventricular y una mayor velocidad poseyección (VPE) aórtica en los QTL sintomáticos, comparados con los asintomáticos. En ese estudio las regiones basales y medio-septales tuvieron mayor VPE, lo que expresa dispersión de la contracción ventricular. En los casos con intervalos QT más prolongados y en las formas mixtas, la duración de la contracción fue mayor.
Con las técnicas ecocardiográficas de Strain circunferencial y longitudinal, Hauggaa demostró mayor duración de la contracción en la región subendocárdica que en la mesomiocárdica de los pacientes con SQTL sintomáticos, comparados con los asintomáticos y los normales.19 Esta dispersión mecánica transmural de la contracción ventricular, evidencia la existencia de un trastorno orgánico asociado con los SQTL congénitos.
MECANISMOS FISIOPATOLÓGICOS Y MOLECULARES
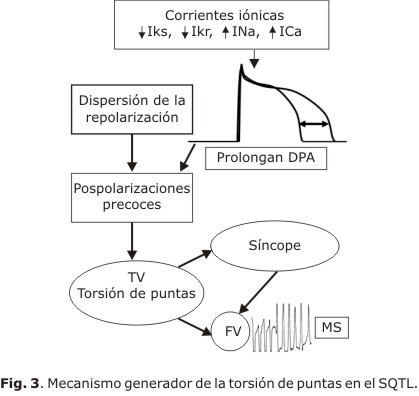
La repolarización ventricular es un proceso no homogéneo, en el que intervienen múltiples factores; aún en el corazón aparentemente sano, las diferencias en la duración de los potenciales de acción y la heterogeneidad de la repolarización, pueden ser los responsables de la aparición de arritmias ventriculares malignas. Ya en 1964 Han y Moe20 habían señalado la relación entre la no homogeneidad de la repolarización y la aparición de arritmias ventriculares. Ellos demostraron que la diferencia entre los períodos refractarios adyacentes podía generar arritmias, incluso fibrilación ventricular.
La prolongación del intervalo QT se asocia con un aumento de la refractariedad y la aparición de los pospotenciales desencadenantes de arritmias ventriculares;21 sin embargo, la heterogeneidad de la repolarización expresada por el incremento de la dispersión del QT, ha sido identificada como el principal sustrato arritmogénico de esta canalopatía. Day y otros,22 en 1990, definen la dispersión de la repolarización ventricular, como la diferencia entre el QT máximo y mínimo en un ECG de 12 derivaciones, pero este recurso ha resultado poco preciso.
Desde hace 2 décadas, se ha utilizado la tesis propuesta por Antzelevitch23 para argumentar la dispersión transmural como índice de dispersión de la repolarización ventricular (dpr), basada en las diferencias electrofisiológicas y funcionales de 3 tipos de células: epicárdicas, M y endocárdicas. Las células M, aunque histológicamente son similares al resto, desde el punto de vista electrofisiológico y farmacológico se comportan como un híbrido entre las células de Purkinje y las ventriculares, lo que determina su respuesta diferente ante determinadas circunstancias. Por ejemplo: el PA en las células M, a diferencia de las células epicárdicas y endocárdicas, se prolonga con la aparición de pospotenciales precoces, en presencia de bloqueadores de las corrientes rectificadoras IKr;21 un hecho similar sucede en presencia de pausas, bradicardias o con el uso de determinados fármacos o trastornos electrolíticos.24,25 Este comportamiento de las células M, origina el aumento de la dispersión transmural de la repolarización (dptr) y constituye el sustrato para el inicio de las taquicardias ventriculares (TV) polimórficas (fig. 2), en específico torsión de puntas (tdp).21,23
En el SQTL, no todas las arritmias ventriculares malignas tienen el mismo mecanismo de origen. En el QTL1 la descarga adrenérgica puede ocasionar acortamiento del PA epicárdico y aumentar la dpr con relación al meso-miocárdico.26 En el QTL2 la dpr es secundaria a la prolongación del PA al nivel de las células M; tanto en el LQT1 como el LQT2 se generan pospotenciales precoces,26 mientras que en el QTL7, la pérdida de la función de las corrientes IK1 en la fase terminal del PA, prolonga la repolarización y produce pospotenciales tardíos.27
La prolongación del PA, al nivel celular, promueve las corrientes ICaL con sobrecarga de Ca2+ y activación de las corrientes activadoras Na+/Ca2+ en las fase 2 y 3 del PA, con la generación de los pospotenciales repetitivos que con frecuencia se autolimitan. Este mecanismo resulta insuficiente para la perpetuación de la arritmia que debe encontrar las condiciones electrofisiológicas de heterogeneidad en el tejido vecino, para que se origine el bloqueo unidireccional y zonas de conducción lenta que faciliten la reentrada.26 El origen y perpetuación de la tdp pueden tener mecanismos diferentes.
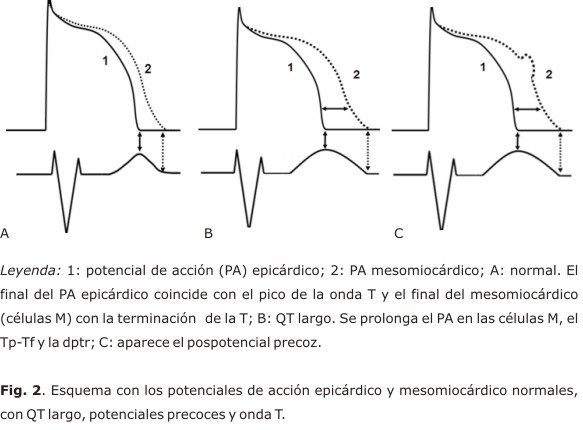
La onda T del ECG periférico, reúne importante información sobre la repolarización ventricular. De acuerdo con los reportes de Antzelevitch,21,23 el PA epicárdico es más corto que el resto y su terminación coincide con la punta de la onda T (Tp), mientras que el miocárdico (células M) concluye con el final de
En los últimos años, se han publicado algunos artículos que rebaten los criterios expuestos y discuten la terminación precoz PA epicárdico, el Tp-Tf como origen de la dptr e incluso, la existencia de las células M.
En el año 2005, Xia y otros,29 publicaron los resultados de un estudio con cerdos, donde obtuvieron registros de PA monofásicos en múltiples sitios de las regiones epicárdicas y endocárdicas de ambos ventrículos, con un sistema de mapeo Carto; ellos midieron la dpr, el Tp y Tf y encontraron que el final del PA epicárdico no coincide con el pico de la onda T, y sí con el tiempo total de repolarización.
En diversas publicaciones, Janse, Coronel y Opthof,30-32 documentan que: el PA epicárdico concluye después el endocárdico, el Tp-f no se relaciona con la dispersión transmural de la repolarización, aunque es un índice de repolarización total y no hay una zona medio-miocárdica (M) con retardo en la repolarización en el corazón intacto; a esto se añade una interrogante sobre el papel arritmogénico que pudieran tener las células M, incluso dudas sobre su existencia.33
La complejidad de la repolarización supera la dispersión transmural, por la no uniformidad en la recuperación que existe entre ápex/base, ventrículo derecho/ventrículo izquierdo y pared anterior/pared posterior.31
Las diferentes preparaciones y modelos utilizados por los distintos investigadores, en la evaluación de los parámetros de la repolarización, parecen haber influido en el debate actual. En el perro normal existe una discordancia entre la polaridad del QRS y la onda T en la mayoría de las derivaciones, a diferencia de lo que sucede con el ECG obtenido de las preparaciones en cuña, en que la polaridad del QRS y T como en el humano, son concordantes.31 Resulta difícil evaluar si estas diferencias entre el corazón íntegro y las preparaciones en cuña, influyen en la adecuada comprensión de la repolarización en el humano. Por otra parte, no todas las preparaciones en cuña se comportan de la misma manera.31
El punto más sensible en esta controversia parece relacionado al valor del Tp-Tf, como índice de dispersión transmural y predictor de arritmias malignas; aunque parece claro el papel de la dpr en la arritmogénesis del SQTL, serán necesario nuevos estudios para aclarar estos aspectos controvertidos.
DIAGNÓSTICO POSITIVO
El SQTL se manifiesta con mareos, síncopes recurrentes, paro cardíaco y MS; la herramienta clave es el ECG con la medición del QT, el QT corregido (QTc) con la frecuencia cardíaca (FC) y los cambios morfológicos del ST y la onda T. Los antecedentes familiares de paro cardíaco o MS, pueden contribuir a orientar el diagnóstico. El test genético, imposible de realizar hasta el momento en nuestro país, permite confirmar el diagnóstico sobre todo en el QTL sin QTL, identificar cada subtipo y estratificar el riesgo.
La medición del QT debe realizarse con el registro a 25 mm/s y amplitud de 10 mm/mv, en las derivaciones II, V5 o V6, desde el inicio del QRS hasta el final de la onda T, en una media de
Las variaciones en la anchura del QRS, como sucede con los bloqueos de rama y uso fármacos antiarrítmicos, modifican la despolarización y pueden también modificar la repolarización, lo que hace inexacta la medición del QT. El cálculo del JT desde
Los valores normales del QTc oscilan entre 350 y 450 milisegundos (ms) en los hombres y entre 360 y 460 ms en las mujeres. Las cifras superiores a 460 ms en los niños y jóvenes de
Las dificultades para el diagnóstico positivo del SQTL con el ECG, fueron bien documentadas por Viskin36 en un estudio con 902 médicos que incluían expertos en QTL, arritmólogos, cardiólogos y no cardiólogos; menos del 50 % de los cardiólogos y del 40 % de los no cardiólogos calcularon el QTc correctamente. La adecuada clasificación de QT largo o corto la realizaron de forma correcta, alrededor del 62 % de los especialistas en arritmias, pero menos del 25 % de los cardiólogos y no cardiólogos.
Para el cálculo del QTc se han utilizado múltiples fórmulas,34 pero es la de Bazett la que ha tenido mayor éxito (QTc= ÖRR);
Algunas pruebas provocativas como el test de ejercicio o de epinefrina pueden contribuir al diagnóstico y a desenmascarar el QTL oculto.37-39 El treadmill puede además, ser útil para predecir el genotipo del SQTL, sobre todo el QTL1.37 La respuesta de la repolarización en la fase de recuperación del ejercicio, puede ser un indicador diagnóstico de SQTL y ayudar identificar los genotipos LQT1 y LQT2.38 La prueba de epinefrina, puede también diferenciar el SQTL de los casos normales y predecir estos genotipos.39
La respuesta del intervalo QT al ligero aumento de FC con la bipedestación, puede ser útil en el diagnóstico del SQTL.40 En un estudio publicado por notables investigadores se demostró que en los pacientes normales al ponerse de pie, el QT se acortó 21 ± 19 ms, mientras que los casos con SQTL, se prolongó 4 ± 34 ms.
El test de Schwartz41 ha sido durante años utilizado por diversos grupos como referencia, para el diagnóstico del SQTL.
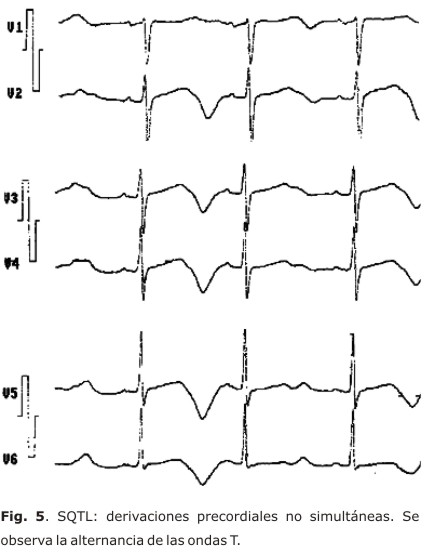
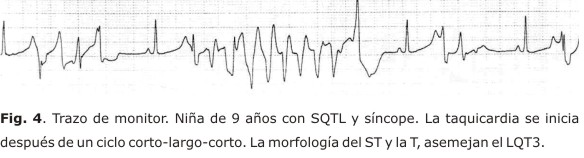
La morfología de la onda T es parte una esencial en el diagnóstico de esta entidad (figs. 4 y 5); puede ser aplanada, bífida, de base ancha o estrecha, alternante (fig. 5), con pico tardío, enlentecimiento en su fase terminal, con onda U prominente o complejo TU. Los 3 subtipos clásicos de SQTL tienen patrones específicos (fig. 1), aunque pueden existir discordancias fenotipo/genotipo secundarias a solapamientos, formas mixtas o variabilidad en la penetración.
La prolongación del QT, la distorsión de
ESTRATIFICACIÓN DE RIESGO
El primer evento arrítmico puede ocurrir a edades tempranas; por lo general el riesgo es mayor en los varones menores de 15 años, sobre todo si son portadores de QTL1. En el sexo femenino parece haber mayor peligrosidad entre los 15 y 40 años.19,42 El síncope, el debut de los síntomas en la infancia, sobre todo en el primer año de vida, el QTc ³ 500 ms, el genotipo (QTL2/QTL3) el sexo femenino y la aparición de eventos arrítmicos a pesar del tratamiento con b-bloqueadores,12,43,44 se consideran predictores de riesgo.
El QTL3 parece ser un importante predictor de eventos arrítmicos.45 Las guías realizadas por
La peligrosidad del SQTL puede ser difícil de determinar debido a la presencia de formas ocultas o latentes, con grados variables de penetración que se hacen evidentes con el uso de fármacos49 (antiarrítmicos o no) u otras situaciones clínicas. En una serie publicada por Yan y otros,50 entre el 10 y 15 % de los SQTL adquiridos por drogas, pueden tener una predisposición genética. La semiología clínica de estos SQTL adquiridos es similar a la que surge en los casos congénitos, con la diferencia que el QT regresa a su estado basal con la suspensión o eliminación del factor desencadenante. Goldenberg y otros,51 en un estudio multicéntrico realizado con 3 386 pacientes, encontraron que el 25 % de los casos era portador de un SQTL oculto y la peligrosidad se incrementaba con la prolongación del QT.
Priori52 documentó que los portadores silentes de SQTL tienen un 20 % de posibilidades de hacerse sintomáticos, pueden trasmitir el QTL hasta al 50 % de su descendencia y que estos últimos son más susceptibles de desarrollar arritmias ventriculares malignas; además, la peligrosidad de la entidad se incrementa con la prolongación del QTc.
Las formas mixtas y los síndromes de superposición con un solo gen pueden modificar la severidad del SQTL congénito; pueden existir enmascaramientos que modifiquen el fenotipo y las manifestaciones clínicas de cada entidad. Por ejemplo: el SCN5A responsable del QTL3 puede asociarse con el síndrome de Brugada y con la fibrilación auricular (FA),53,54 pero el gen es común a otros trastornos como la enfermedad del sistema de conducción, la disfunción sinusal y la parálisis auricular. Otras asociaciones KCNQ1/ FA familiar y variantes del KCNH2,54 pueden también encontrarse enmascarados. Este comportamiento pleiotrópico puede modificar la evolución clínica de la enfermedad, en dependencia del trastorno que predomine, factores genéticos-ambientales y el sustrato específico del paciente.
Las mutaciones pueden también ser compuestas, con más de 1 gen. Un estudio publicado por Westenskow y otros55 con 252 casos, documentó que el 7,9 % tenía 2 variantes de genes y que en el grupo con 2 mutaciones el QTc era más largo, con mayor incidencia de arritmias y síntomas más graves.
En un estudio multicéntrico realizado con la población japonesa, se encontró que los portadores genéticos de QTL1 con mutaciones transmembrana, tenían mayor riesgo de eventos cardíacos y mayor sensibilidad a la estimulación simpática, que los que tenían mutaciones en C terminal.56
El riesgo de eventos cardíacos en los pacientes con SQTL congénito, depende de múltiples factores, y aunque el test genético, en los lugares donde se puede realizar, permite en gran medida tipificar los casos y definir los pacientes con mayor riesgo, alrededor del 30 % de los estudios genéticos convencionales no logran descubrir alguna mutación,16 pueden existir falsos positivos, falsos negativos o dificultades técnicas que no permitan la adecuada tipificación; no obstante, es una prueba de extrema importancia. Las indicaciones del test genético para las canalopatías, han sido recientemente publicadas por un consenso de expertos.57
La dispersión mecánica de la contracción ventricular documentada por Haugaa en los SQTL congénitos con las técnicas de Strain,19 puede resultar un predictor de riesgo; la mayor duración de la contracción ventricular en los pacientes sintomáticos, en relación con los asintomáticos y el grupo control, puede servir para identificar los casos con mayores probabilidades presentar de arritmias malignas.
CONDUCTA
Si la sintomatología en el SQTL se relaciona al deporte, ejercicios, estrés emocional, alarmas u otras situaciones que generen estimulación adrenérgica, es imprescindible modificar el sistema de vida y la eliminación de los posibles desencadenantes, lo que puede incluir la separación del deporte activo o competitivo, el control o reducción del sonido de las alarmas del reloj, timbres u otros como los originados en las discotecas.14,43
Para definir el tratamiento debe descartarse el uso de fármacos, trastornos electrolíticos (hipopotasemia, hipomagnesemia), bradicardias, pausas, disfunción hepática o renal u otros factores que pudieran justificar la prolongación adquirida del QTc, en asociación o no con el SQTL congénito.
Los b-bloqueadores constituyen la primera línea de tratamiento, con una respuesta favorable en el LQT1 y LQT2; en el LQT3 pueden ser inefectivos, incluso tener un efecto perjudicial.14,43,58,60
Los suplementos de potasio y la espironolactona, también pueden ser útiles sobre todo en el LQT2.14,43
La denervación simpática izquierda ha demostrado resultados favorables en el LQT1 refractario a los b-bloqueadores.43
El uso de bloqueadores de los canales de Na como la mexiletina, flecainide y ranolazina pueden tener un efecto beneficioso en los LQT3, pero su uso sin la orientación genética pudiera ser dañino, en caso de asociación con otras entidades como el síndrome de Brugada.14,58,60
El verapamilo, que es también un inhibidor de las corrientes tardías de Na+, pudiera reducir el QTL, la dpr y los pospotenciales en algunos modelos de SQTL.59
Los marcapasos cardíacos son efectivos en las TV pausa-dependientes (intervalos corto-largo-corto), bradicardias o bloqueos aurículo ventriculares con o sin prolongación del intervalo QTc.58,60 Tienen mayor utilidad en el SQT3. Su asociación con β-bloqueadores puede ser útil en los pacientes con alto riesgo.
Los antecedentes de paro cardíaco o MS abortada, síncopes recurrentes a pesar del tratamiento farmacológico o TV sostenidas, constituyen indicación de DAI.58,60 Estos dispositivos pueden ser efectivos como prevención primaria en los pacientes con fuerte historia familiar de MS, intolerancia al tratamiento medicamentoso o QTc ³ 500 ms.58,60
Han transcurrido más de 50 años de las primeras descripciones del SQTL; múltiples estudios y publicaciones se han realizado y avalado por prestigiosos investigadores que respaldados por el desarrollo genético-molecular, han aportado importantes elementos a la compresión de esta compleja entidad; no obstante, todavía quedan incógnitas y controversias que se deberán solucionar, en la medida que lo permita el desarrollo científico técnico y el progresivo ascenso del conocimiento de esta canalopatía, la más antigua descrita.
REFERENCIAS BIBLIOGRÁFICAS
1. Jervell A, Lange-Nielsen F. Congenital deal-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J. 1957;54:59-68.
2. Romano C, Gemme G, Pongiglione R. Rare cardiac arrhythmias of the pediatric age. II. Syncopal attacks due to paroxysmal ventricular fibrillation (presentation of first case in Italian pediatric literature) [Italian]. Clin Pediatr. (
3. Ward OC. A new familial cardiac syndrome in children. J Isr Med Assoc. 1964;54:103-6.
4. Priori SG, Schwartz PJ, Napolitano C, Bianchi L, Dennis A, De Fusco M, et al. A recessive variant of the Romano-Ward long-QT syndrome? Circulation. 1998;97:2420-25.
5. Schwartz PJ. The idiopathic long QT syndrome. The need for a prospective registry. Eur Heart J. 1983;4:529.
6. Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Spazzolini C. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761-67.
7. Moss AJ, Robinson JL. The long QT syndrome. Circulation 2002;105:784-86.
8. Hedley PL, Jorgensen P, Schlamowitz S, Wangari R, Moolman-Smook J, Brink PA, et al. The genetic basis of long and short QT syndromes: A mutation update. Human Mutation. 2009;30:1486-1511.
9. Baruteau A-E, Baruteau J, Joomye R, Martins R, Treguer F, Baruteau R, et al. Role of congenital Long-QT in unexplained sudden infant death: proposal for an electrocardiographic screening in relatives. Eur J Pediatr. 2009;168:771-77.
10. Grant AO. Cardiac ion channels. Circ. Arrhythmia Electrophysiol. 2009;2:185-94.
11. Ackerman MJ, Mohler PJ. Defining a new paradigm for human arrhythmias syndromes: Phenotypic manifestations of gene mutations in ion channels-and transporter-associated proteins. Circ Res. 2010;107:457-65.
12. Tester DJ, Ackerman MJ. Genetic testing for potentially lethal, highly treatable inherited cardiomyopathies in clinical practice. Circulation. 2011;123:1021-37.
13. Yang Y, Yang Y, Liang B, Liu J, Li J, Grunnet M, et al. Identification of Kir3.4 mutation in congenital long QT syndrome. Am J Hum Gen. 2010;86:872-80.
14. Ruan Y, Liu N, Napolitano C, Priori SG. Therapeutic strategies for long-QT syndrome. Does the molecular substrate matter? Circ Arrhythmia Electrophysiol. 2008;1:290-7.
15. Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866-74.
16. Rossenbacker T, Priori SG. Nuevas perspectivas en el síndrome de QT largo. Rev Esp Cardiol. 2007;60:675-82.
17. Haugaa KH, Edvardsen T, Leren TP, Gran JM, Smiseth OA, Amlie JP. Left ventricular mechanical dispersion by tissue Doppler imaging: a novel approach for identifying high-risk individuals with long QT syndrome. Eur Heart J. 2009;30:330-37.
18. Nador F, Beria G, De Ferrari GM, Stramba-Badiale M, Locati EH, Lotto A, et al. Unsuspected echocardiographic abnormality in the long Q-T syndrome: diagnostic, prognostic, and pathogenetic implications. Circulation. 1991;84:1530-42.
19. Haugaa KH, Amlie JP, Berge KE, Leren TP, Smiseth OA, Edvardsen T, et al. Transmural differences in myocardial contraction in Long-QT syndrome. Mechanical consequences of ion channel dysfunction. Circulation. 2010;122:1355-63.
20. Han J, Moe GK. Nonuniform recovery of excitability in ventricular muscle. Circ Res. 1964;14:44-60.
21. Antzelevitch C. Role of spatial dispersion of repolarization in inherited and acquired sudden cardiac death syndromes. Am J Physiol Heart Circ Physiol. 2007;293:2024-38.
22. Day CP, Mc Comb JM,
23. Antzelevitch C, Sicouri S, Litovsky SH, Lukas A, Krishnan SC, Di Diego JM, et al. Heterogeneity within the ventricular wall: Electrophysiology and pharmacology of epicardial, endocardial and M cells. Cir Res. 1991;69:1427-49.
24. Liu J, Laurita KR. The mechanism of pause-induced torsade de pointes in long QT syndrome. J Cardiovasc Electrophysiol. 2005;16:981-87.
25. Childers Rory. Torsades: adjacent and triggering electrocardiographic events. J Electrocardiol. 2010:43:515-23.
26. Rosembaum DS. Is long QT syndrome a disease of abnormal mechanical contraction? Circulation. 2010;122:1353-54.
27. Shimizu W, Horie M. Phenotypic manifestations of mutations in genes encoding subunits of cardiac potassium channels. Circ Res. 2011;109:97109.
28. Panikkath R, Reinier K, Uy-Evanado A, Teodorescu C, Hattenhauer J, Mariani R, et al. Prolonged T peak-to-T end interval on the resting ECG is associated with increased risk of sudden cardiac death. Circ Arrhythm Electrophysiol. 2011;4:441-47.
29. Xia Y, Liang Y, Kongstad O, Holm M, Olsson B, Yuan Sh. Tpeak-Tend as an index of global dispersion of ventricular repolarization: evaluations using monophasic action potential mapping of the epi-and endocardium in swine. J Inter Card Electrophysiol. 2005;14:79-87.
30. Opthof T, Coronel R, Wilms-Schopman FJG, Plotnikov AN, Shlapakova IN, Danilo P, et al. Dispersion of repolarization in canine ventricle and the electrocardiographic T wave: Tp-e interval does not reflect transmural dispersion. Heart Rhythm. 2007;4:341-48.
31. Opthof T, Coronel R, Janse MJ. Is there a significant trasmural gradient repolarization time in the intact heart? Circulation 2009;2:89-96.
32. Janse MJ, Coronel R, Opthof T. Counterpoin: M cells do not have a functional role in the ventricular myocardium of intact heart. Heart Rhythm. 2011;8:934-37.
33. Morita Sh T, Zipes DP, Morita H, Wu J. Analysis of action potentials in the canine ventricular septum: No phenotypic of M cell. Cardiovasc Res. 2007;74:96-103.
34. Goldenberg I, Moss AJ, Zareba W. QT interval: How to measure it and what is "normal". J Cardiovasc Electrophysiol. 2006;17:333-36.
35. Viskin S. The QT interval: Too long, to short or just right. Heart Rhythm. 2009;6:711-15.
36. Viskin S, Rosovski U, Sands AJ, Chen E, Kistler PM, Kalman JM, et al. Inaccurate electrocardiographic interpretation of long QT: The majority of physicians cannot recognize a long QT when they see one. Heart Rhythm. 2005;2:569-74.
37. Wong JA, Gula LJ, Klein GJ, Yee R, Skanes AC, Krahn AD. Utility of tradmilltesting in identification and genotype prediction in Long-QT Syndrome. Circ Arrhythm Electrophydiol. 2010;3:120-25.
38. Chattha IS, Sy RW, Yee R, Gula LJ, Snakes AC, Klein GJ, et al. Utility of the recovery electrocardiogram aftere exercise and genotyping of long QT syndrome. Heart Rhythm. 2010;7:906-11.
39. Hekkala AM, Swan H, Vittasalo M, Vaananem H, Toivonen L. Epinephrine bolus test in detectin long QT syndrome mutation carriers with indeterminable electrocardiographic phenotype. Ann Noninvasive Electrocardiol. 2011;16:172-9.
40. Viskin S, Postema PG, Bhuiyan ZA, Rosso R, Kalman JM, Vorha JK, et al. The respons of the QT interval to brief tachycardia provoked by standing. A bedside test for diagnosing long QT syndrome. J Am Coll Cardiol. 2010;55:1955-61.
41. Schwartz PJ, Moss A, Vincent GM, Crampton RS. Diagnostic criteria for long QT syndrome. Circulation. 1993;88:782-84.
42. Locati EH, Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Lehmann MH, et al. Age- and sex-related differences in clinical manifestations in patients with congenital long-QT syndrome: findings from the international LQTS registry. Circulation. 1998;97:2237-44.
43. Moss AJ, Goldenberg I. Importance of knowing the genotype and the specific mutation when managing patients with long-QT syndrome. Circ Arrhythm Electrophysiol. 2008;117:219-26.
44. Sauer AJ, Moss A, McNitt S,
45. Goldenberg I, Moss AJ, Bradley J, Polonski S, Peterson DR, McNitt S, et al. Long-QT after age 40. Circulation. 2008;117:2192-01.
46. Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death) developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society Europace. 2006;8:746-837.
47. Schwartz PJ, Spazzolini C, Priori SG, Crotti L, Vicentini A, Landolina M, et al. Who are the long-QT syndrome patients who receive an implantable cardioverter-defibrillator and what happens to them? Data from the European Long-QT syndrome implantable cardioverter-defibrillator (LQTS ICD) registry. Circulation. 2010;122;1272-82.
48. Schwartz PJ, Spazzolni C, Crotti L. All LQT3 patients need an ICD: True o false? Heart Rhythm. 2009;6:113-20.
49. Gupta A, Lawrence AT, Krishnan K, Kavinsky CJ, Throman R. Currents concepts in the mechanisms and management of drug-induced QT prolongation and torsade de points. Am Heart J. 2007;153:891-9.
50. Yang P, Kanki H, Drolet B, Yang T, Wei J, Viswanathan PC, et al. Allelic variants in long-QT disease genes in patients with drug-associated torsades de pointes. Circulation. 2002;105:1943-8.
51. Goldenberg I, Horr S, Moss AJ, Lopes CM, Barsheshet A, McNitt S, et al. Risk for life-threatening cardiac events in patients with genotype-confirmed long-QT syndrome and normal-range corrected QT intervals. J Am Coll Cardiol. 2011;57:51-9.
52. Priori SG. Inherited arrhythmogenic diseases: the complexity beyond monogenic disorders. Circ Res. 2004;94:140-5.
53. Remme CA, Wilde AAM, Bezzina CR. Cardiac sodium channel overlap syndromes: different faces of SCN5A mutations. Trends Cardiovasc Med. 2008;18:78-87.
54. Crotti L. Pleiotropic mutations in ion channels: What lies behind them? Heart Rhythm. 2011;8:56-7.
55. Westenskow P, Splawski I, Timothy KW,
56. Shimizu W, Horie M, Ohno S, Takenaka K, Yamaguchi M, Shimizu M, et al. Mutation site-specific differences in arrhythmic risk and sensitivity to sympathetic stimulation in LQT1 form of congenital long QT syndrome: multicenter study in Japan. J Am Coll Cardiol. 2004;44:117-25.
57. Ackerman MJ, Priori SG, Willems S, Berul Ch, Brugada R, Calkins H, et al. HRS/EHRA Expert consensus statements on the state of genetic testing fon channelophaties and cardiomyopathies. Heart Rhythm. 2011;8:1308-39.
58.
59. Aiba T, Shimizu W, Inagaki M, Noda T, Miyoshi S, Ding WG, et al. Cellular and ionic mechanism for drug-induced long-QT syndrome and effectiveness of Verapamil. J Am Coll Cardiol. 2005;45:300-7.
60. Epstein AE, DiMarco JP, Ellenbogen KA, Mark Estes NA, Freedman EA, Gettes LS, et al. ACC/AHA/HRS 2008 Guidelines for device-based therapy of cardiac rhythm abnormalities: A report of American College of Cardiology/American Heart Association Task force on practice guidelines (Writing Committee to revise ACC/AHA/NASPE 2002 guideline update for implantation of cardiac pacemaker and antiarrhythmia devices): Developed in collaboration with the American Association for Thoracic Surgery and Society of Thoracic Surgeons. Circulation. 2008;117:e350-8.
Recibido: 30 de marzo del 2012.
Aprobado: 1 de junio del 2012.
Dr. Roberto Zayas Molina. Instituto de Cardiología y Cirugía Cardiovascular.

{kind=link}
{kind=link}