










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Investigaciones Biomédicas
versión impresa ISSN 0864-0300
Rev Cubana Invest Bioméd vol.31 no.2 Ciudad de la Habana abr.-jun. 2012
REVISIÓN
Síndrome de QT corto
Short QT syndrome
Dra. Marleny Cruz Cardentey
Hospital Clinicoquirúrgico "Hermanos Ameijeiras".
RESUMEN
El síndrome de QT corto es una canalopatía hereditaria caracterizada por un anormal acortamiento del intervalo QT (IQT), por un riesgo incrementado para el desarrollo de fibrilación auricular y/o arritmias ventriculares malignas y por la ausencia de cardiopatía estructural. Es una enfermedad heterogénea y se han identificado mutaciones en los genes codificadores de los canales de potasio y de calcio. Un incremento en las corrientes neta de salida de potasio o una disminución en al entrada de calcio favorecen el acortamiento heterogéneo de la repolarización ventricular. La marcada abreviación de la longitud de onda del circuito es un factor arritmogénico adicional. El curso clínico oscila desde formas asintomáticas hasta fibrilación auricular paroxística o permanente, síncope, arritmias ventriculares y muerte súbita. El electrocardiograma muestra IQT 220-360 ms, ondas T altas y puntiagudas, prolongación del intervalo pico-final de la onda T e IQT rígido. Es poco frecuente, pero importante por el riesgo elevado de muerte súbita, que en ocasiones puede ser el debut. Puede presentarse solapado al síndrome de Brugada y a la repolarización precoz. El diagnóstico precisa excluir las causas secundarias que acortan el IQT y la no identificación de una mutación no lo excluye. La estimulación eléctrica programada tiene pobre valor diagnóstico y pronóstico. En los sujetos con muerte súbita abortada o con arritmias ventriculares con compromiso hemodinámica, el desfibrilador es la terapéutica de elección. La quinidina es una opción terapéutica alternativa.
Palabras clave: síndrome de QT corto, muerte súbita, canalopatías.
ABSTRACT
The short QT syndrome is an inherited channelopathy characterized by an abnormal shortening of the QT interval (QTI), an increased risk of developing atrial fibrillation and/or malignant ventricular arrhythmias, and the absence of structural heart disease. It is a heterogeneous disease and mutations have been identified in the genes encoding potassium and calcium channels. An increase in potassium net efflux or a decrease in calcium influx facilitate the heterogeneous shortening of ventricular repolarization. A marked shortening of the wavelength of the circuit is an additional arrhythmogenic factor. The clinical course ranges from asymptomatic forms to paroxysmal or permanent atrial fibrillation, syncope, ventricular arrhythmias and sudden death. The ECG shows QTI 220-360 ms, high and sharp T waves, prolongation of the final peak interval of the T wave, and QTI drive. It is a rare disease whose importance lies in the high risk of sudden death, which may sometimes be its debut. It may overlap Brugada syndrome and early repolarization. Diagnosis requires excluding secondary causes of QTI shortening. Failure to identify a mutation does not exclude it. Programmed electrical stimulation has a low diagnostic and prognostic value. Defibrillation is the therapy of choice for patients with aborted sudden death or ventricular arrhythmias with hemodynamic compromise. Quinidine is an alternative therapeutic option.
Key words: short QT syndrome, sudden death, channelopathies.
INTRODUCCIÓN
El síndrome de QT corto (SQTC) es un desorden eléctrico primario del corazón, de carácter hereditario y caracterizado por un anormal acortamiento del intervalo QT (IQT) en el electrocardiograma de superficie y por un riesgo incrementado para el desarrollo de fibrilación auricular y/o arritmias ventriculares malignas. Esta entidad ha tenido una reciente incorporación a la lista de las canalopatías responsables de la muerte súbita cardíaca en individuos con corazón estructuralmente normal al nivel macroestructural.1,2
El posible potencial arritmogénico del acortamiento del IQT fue propuesto inicialmente por Algra y otros3 en el año 1993. Estos autores reportaron en los sujetos con un IQT< 400 ms un riesgo relativo de muerte súbita cardíaca 2,4 mayor que en los que mostraban un IQT normal. Viskin y otros4 también describieron una anormal reducción del IQT en los pacientes con fibrilación ventricular idiopática. La abreviación del IQT reportada antes y después de los episodios de taquicardia y fibrilación ventricular,5,6 es otro elemento que fundamenta el probable valor patológico de un IQT corto. Curiosamente, esta alteración electrocardiográfica es un hallazgo normal en especies como el canguro. Sin embargo, en estos animales se recoge una alta incidencia de muerte súbita.7
No obstante, recientes estudios en poblaciones sanas han cuestionado la significación arritmogénica del acortamiento del IQT. Según estas series, su real incidencia es extremadamente baja (0,01-0,5 %), no se asocia con un incremento del riesgo de muerte súbita cardíaca en un seguimiento entre 8 y 30 años y puede representar una parte de la curva normal de distribución de la población sana.8,9
La abreviación de la repolarización ventricular parece no ser suficiente para crear el sustrato arritmogénico. El SQTC precisa de un incremento significativo en la dispersión de la repolarización, la cual instaurará las bases celulares para la génesis de la reentrada. Estas distinciones nos permiten comprender por qué la abreviación del IQT es altamente arritmogénico en algunos individuos y en otros no.
BASES GENÉTICAS
EL SQTC es una enfermedad heterogénea. Hasta la fecha se han identificado mutaciones en 5 genes diferentes, todas con un patrón de herencia autosómica dominante. Según el orden cronológico en que han sido descritas se han nombrado como SQTC1 hasta SQTC5 (tabla). Excepto en el SQTC2, que se reportó en un paciente aislado, en las restantes formas de SQTC las mutaciones se han comprobado en más de un miembro de una misma familia.
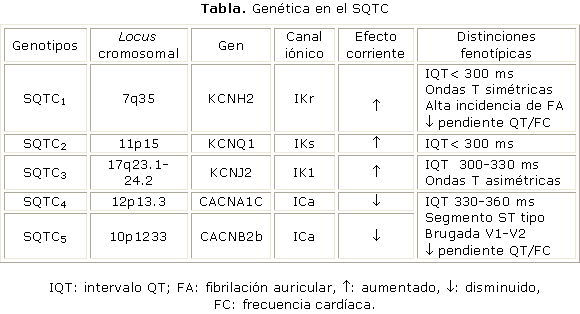
Mutación en KCNH2-SQTC1
El gen KCNH2 codifica la subunidad alfa del componente rápido de la corriente tardía rectificadora de potasio (IKr). Brugada y otros10 identificaron 2 mutaciones en familias no relacionadas; en ambas existe una sustitución de aspargina por lisina en el codón 588 del poro del canal. La mutación suprime la inactivación del canal, con un incremento en la corriente neta de salida de potasio. La ganancia de función del IKr genera un acortamiento en la duración del potencial de acción y en el período refractario auricular y ventricular, justificación para la alta asociación de arritmias ventriculares con fibrilación auricular reportada en este subtipo genético.
Mutación en KCNQ1-SQTC2
El gen KCNQ1 codifica una proteína del componente lento de la corriente rectificadora tardía de potasio (IKs). Bellocq y otros11 identificaron la primera mutación del gen en un hombre de 70 años resucitado de un episodio de fibrilación ventricular y con un IQT de 290 ms posreanimación. Más recientemente, se describió una nueva mutación en una recién nacida de 38 semanas con fibrilación auricular con respuesta ventricular lenta e IQT corto. Las mutaciones producen una hiperfunción del canal IKr, acortándose la refractariedad auricular y ventricular.12
Mutación en KCNJ2- SQTC3
En el SQTC3 se produce una mutación en el gen que codifica al canal rectificador de potasio IK1. Priori y otros13 identificaron la primera mutación en una niña de 5 años y en su padre. Ambos pacientes mostraban un IQT no excesivamente corto (315 y 320 ms, respectivamente) y una onda T asimétrica con ascenso lento y fase terminal rápida. Su padre refería además cuadros presincopales precedidos de palpitaciones. Los efectos electrofisiológicos del canal mutante se traducen en una ganancia de su función, con aceleración de la repolarización tardía y acortamiento del período refractario.
Mutación en SQTC4- SQTC5
Recientemente se describió la combinación fenotípica de un IQT corto con un ST tipo síndrome Brugada en V1-V2, basal o tras la administración de ajmalina. El screening genético en 3 familias con las citadas distinciones fenotípicas reveló mutaciones en los canales lentos de calcio. Todas las mutaciones testadas implican una marcada pérdida de la función del canal. El IQT en estos 2 subtipos, no suele estar marcadamente acortado (330-360 ms) en comparación con las restantes formas de SQTC. Muestran además ondas T altas y puntiagudas, así como una pérdida de la adaptación del IQT a los cambios de la frecuencia cardíaca, similar a como sucede en el SQTC1.14
A pesar de un minucioso cribado genético, hay pacientes sintomáticos con un IQT corto en los que no se ha identificado la mutación responsable. Al igual que en el síndrome de QT largo, la presentación clínica, los desencadenantes del episodio arrítmico y las opciones terapéuticas en el SQTC, pudieran estar determinadas por la mutación genética. Se necesitarán estudios futuros para identificar nuevos sitios mutantes, así como para establecer una correlación genotipo-fenotipo en esta entidad.
BASES CELULARES EN EL SQTC
Un incremento en las corrientes neta de salida de potasio o una disminución en al entrada de calcio favorecen el acortamiento de la repolarización ventricular y del IQT. Estudios experimentales sugieren que la abreviación del potencial de acción en el SQTC es heterogénea, con acortamiento preferencial en el epicardio, endocardio y células de Purkinje, situación que genera un incremento de la dispersión transmural de la repolarización ventricular, que a su vez actuará como sustrato de la reentrada al favorecer el bloqueo unidireccional.15,16 En los pacientes con SQTC el intervalo pico-final de la onda T y la relación pico-final de la onda T/QT están incrementadas.17 Significativamente esta correlación es más ostensible en los pacientes sintomáticos.18
Una marcada abreviación de la longitud de onda del circuito, secundario al acortamiento de la refractariedad, es un factor adicional que favorece el mantenimiento de la reentrada en esta miopatía eléctrica.16
Los episodios de taquicardia ventricular polimórficas originados en los pacientes con SQTC, generalmente son desencadenados por una extrasístole ventricular con intervalo de acoplamiento muy corto.19 Se especula que la génesis del complejo ventricular prematuro pudiera estar en relación con una reentrada en fase 2 o con posdespolarizaciones precoces al final de la fase 3 del potencial de acción.20
Estudios recientes demostraron que la actividad eléctrica en el SQTC termina antes de que culmine la contracción mecánica, generando una disociación electro-mecánica.21 Similar evento ha sido reportado previamente en canguros. En esta especie se ha observado que la ocurrencia de una extrasístole re-excitará el miocardio cuando aún no ha culminado la actividad mecánica del latido precedente, creando un estado de tétanos incompleto.22
PRESENTACIÓN CLÍNICA
La presentación clínica es altamente variable, aún en miembros de una misma familia. El curso clínico oscila desde formas asintomáticas hasta fibrilación auricular paroxística o permanente, síncope, arritmias ventriculares y muerte súbita. El debut de la entidad ha sido reportado tan precozmente como en el primer año de vida (pudiendo estar en relación con la muerte súbita del infante) y tan tardío como a los 80 años. Una fuerte historia familiar de muerte súbita y/o fibrilación auricular es un hallazgo común en estos pacientes.1,23,24
Giustetto y otros25 muestran la serie más amplia con 29 pacientes. El 62 % de ellos es sintomático, la muerte súbita es el síntoma más frecuente (34 %) y representa el debut de la enfermedad en el 28 % de los afectados. Las palpitaciones (31 %) y el síncope (24 %) son los restantes síntomas más habituales. La fibrilación auricular aislada como síntoma de inicio de la entidad se presenta en el 17 % de los pacientes. Los episodios arrítmicos han sido reportados durante el reposo, el ruido, el ejercicio y la actividad cotidiana.
La estimulación eléctrica programada en el SQTC tiene una baja sensibilidad para la inducción de arritmias ventriculares (50 %). La no inducción en el laboratorio no excluye el riesgo de desarrollar un evento futuro. Fundamentado por su pobre valor diagnóstico y pronóstico no se utiliza de forma extensiva en esta entidad.25
Caracerísticas electrocardiográficas
1. Acortamiento del IQT, con un rango entre 220-360 ms. En el SQT1-2 el IQT está marcadamente abreviado (> 300 ms), mientras que en el SQTC4-5 el acortamiento no es tan acentuado (330-360 ms).10,11,14 Las abreviaciones del IQT pueden ser transitorias, dándole valor diagnóstico a la monitorización de Holter y a las secuencias eléctricas (fig.).
2. Ondas T altas, puntiagudas y con segmento ST prácticamente ausente. La existencia de alteraciones en la onda T, a pesar de ser un hallazgo común en el SQTC, no parece guardar relación con el potencial arritmogénico de la entidad. En el SQTC1-2 las ondas T son simétricas, mientras que en el SQTC3 son asimétricas, con un ascenso lento y una rama descendente rápida. La apariencia peculiar de la onda T en el SQTC3 puede estar en relación con la aceleración súbita del final de la repolarización ventricular, proceso mediado por los canales IK1.13,24,25
3. Secundario al acortamiento no homogéneo de la repolarización ventricular se prolonga el en la duración del intervalo pico-final de la onda T, así como la relación del intervalo pico final de la onda T-intervalo QT.15,17
4. IQT rígido y con una pobre modificación tras los cambios de la frecuencia cardíaca. La pérdida de la adaptación del IQT a los cambios de frecuencia hacen que la curva de la relación IQTD RR muestre una menor pendiente.23,26
5. En el SQTC4 y SQTC5 además del acortamiento del IQT se observa una elevación del segmento ST tipo Brugada en V1-V2, basal o tras la administración de ajmalina.14
6. En los sujetos con SQTC existe una alta incidencia de repolarización precoz y su hallazgo se asocia con el desarrollo de eventos arrítmicos. Recientes trabajos sugieren que la repolarización precoz pudiera ser un marcador de riesgo de muerte súbita cardíaca en los pacientes con SQTC.27
7. Acortamiento del intervalo punto J-pico de la onda T. Abreviación significativamente más acentuada en los pacientes con SQTC sintomáticos.18
El SQTC puede presentarse como una canalopatía aislada o solaparse con otras miopatías eléctricas (síndrome de Brugada y repolarización precoz).14,17 El enmascaramiento de las enfermedades de los canales iónicos, sugiere la existencia de sustratos genéticos, moleculares, iónicos, electrofisiológicos, arritmogénicos y terapéuticos comunes.
Algunas consideraciones para la corrección del IQT
La corrección del IQT mediante la utilización de las fórmulas convencionales en el SQTC, no es fidedigna. A frecuencias cardíacas elevadas el IQT puede falsamente normalizarse, situación de particular importancia el la edad pediátrica, donde la frecuencia cardíaca normal en reposo es superior a 100 latidos por minuto (lpm). Por lo tanto, es recomendable corregir el IQT a frecuencias cardíacas no superiores a 85 lpm.28
La utilización de la fórmula de Bazett para calcular el IQT corregido en varios estudios poblacionales a gran escala, tiende a subestimar el IQT a frecuencias cardíacas bajas y a sobreestimarlo a frecuencias altas. Catalogando erradamente un IQT corto en el 20 % de la población adulta sana, en especial si el elecrocardiograma fue registrado durante una bradicardia sinusal.28,29
Mediante la medición del IQT en 14 379 individuos sanos, Rautaharju y otros,30 proponen un nuevo método para estimar el IQT ajustado a la frecuencia cardíaca. Los autores establecen una fórmula para calcular el IQT predecible (IQTp)= 656 / (1+ frecuencia cardíaca / 100). En este estudio, un IQT< 88 % del IQTp equivale a 2 desviaciones estándares de la media y representa solo el 2,5 % de la población adulta sana. Por ejemplo, para una frecuencia cardíaca de 60 lpm el IQTp es de 410 ms; el 88 % del IQTp es 360 ms. Valores inferiores a 360 ms deberán ser considerados como IQT corto. Debido a la rigidez del IQT en el SQTC, muchos autores prefieren este método a la convencional fórmula de Bazett.20
Diagnóstico del SQTC
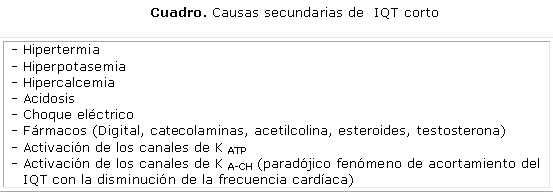
Hasta la fecha aparecen publicados en la literatura de habla inglesa 61 casos de SQTC.31 Para establecer el diagnóstico de la entidad se precisa: IQT corto < 360 ms en ausencia de causas secundarias (cuadro), más la existencia de alguna de las siguientes condiciones:20,24,31,32
1. Fibrilación auricular y/o fibrilación ventricular (documentadas o con síntomas en relación con estas arritmias).
2. Antecedentes familiares de muerte súbita y/o SQTC.
La existencia de un IQT corto aislado, en ausencia de síntomas o historia familiar, debe considerarse un signo electrocardiográfico de IQT corto y no el síndrome en sí, tal como sucede con el síndrome de Brugada y el electrocardiograma tipo Brugada. No debe cometerse el error de diagnosticar una entidad fuera del contexto clínico.32
A todos los pacientes se le debe realizar: electrocardiogramas seriados en reposo y en esfuerzo, monitorización de Holter, así como ecocardiograma para descartar la existencia de una cardiopatía estructural. El test genético debería en potencia realizarse a todos los pacientes diagnosticados, pero por lo engorroso y altamente costoso, solo se efectúa en escasos centros del mundo. Es importante recalcar que el SQTC es una enfermedad heterogénea y que la no identificación de una mutación genética no excluye su diagnóstico. El electrocardiograma, el cuadro clínico y la historia familiar son suficientes para diagnosticar la entidad.
TRATAMIENTO
Desfibrilador automático implantable (DAI)
La alta incidencia de muerte súbita cardíaca por arritmias ventriculares malignas en el SQTC recomienda la indicación de un DAI.33 Debido a su reciente descripción, los registros e investigaciones disponibles son insuficientes para establecer la estratificación de riesgo de muerte súbita en esta miopatía eléctrica. La sensibilidad de la estimulación eléctrica programada para la inducción de arritmias ventriculares en el SQTC es de alrededor del 50 % y la no inducibilidad no excluye el riesgo de desarrollar un evento futuro.19,25 Los estudios genéticos igualmente son limitados en la estratificación de riesgo en esta canalopatía. Consecuentemente, la indicación del DAI en la prevención primaria de muerte súbita en esta entidad no es aconsejable, aunque deberá individualizarse en los pacientes con una fuerte carga familiar. En los sujetos con muerte súbita abortada o con arritmias ventriculares con compromiso hemodinámico, la indicación de un DAI es la opción terapéutica de elección.20,25
La terapia inapropiada del dispositivo secundario al sobresensado de la onda T, es una complicación frecuente en los pacientes con SQTC y DAI. El uso de quinidina y la reprogramación del desfibrilador, son suficientes para prevenir el choque inadecuado.34
Tratamiento farmacológico
Aunque el DAI es la principal opción terapéutica en el SQTC, el tratamiento con fármacos es una terapia igualmente útil, aislada o en asociación con el dispositivo. Se recomienda su empleo en las siguientes situaciones: a) edad pediátrica donde el implante de una DAI pudiera ser problemático; b) negativa al implante del DAI; c) prevención primaria de muerte súbita en los pacientes asintomáticos; d) prevención de las descargas inapropiadas del dispositivo por sobresensado de la onda T.20
Las investigaciones existentes donde se evalúa la efectividad de los fármacos antiarrítmicos en el SQTC son limitadas, con muestras pequeñas y confinadas al SQTC1. La quinidina ha sido la única droga que ha mostrado beneficios: prolonga el IQT y los períodos refractarios, restaura la relación IQT/frecuencia cardíaca, disminuye la inducción de fibrilación ventricular y reduce el voltaje de la onda T y por consiguiente, las terapias inapropiadas del DAI.35
En su mecanismo de acción, la quinidina bloquea múltiples canales de potasio (Ito, IK1, IKr, IKs). En potencia, este fármaco pudiera ser igualmente efectivo para los restantes subtipos genéticos del SQTC, especialmente para el SQTC4 y SQTC5, donde el bloqueo de las corrientes Ito de potasio modifica el sustrato y el disparador en el síndrome de Brugada.14
Schimpf y otros,36 han reportado en un estudio reciente la eficacia clínica de la disopiramida en 2 mujeres con SQTC1. El uso de la citada droga prolongó el IQT y restauró el período refractario ventricular. La amiodarona igualmente prolongó el IQT en 2 pacientes con SQTC de genotipo desconocido.37
En los pacientes con fibrilación auricular aislada, como forma de presentación del SQTC, la propafenona ha mostrado efectividad en prevenir los paroxismos de la taquiarritmia en un seguimiento de 2 años. Sin embargo, este fármaco no modifica la duración del IQT.38 La quinidina es igualmente eficaz en el manejo de la fibrilación auricular solitaria.
REFERENCIAS BIBLIOGRÁFICAS
1. Gussak I, Brugada P, Brugada J, Wright RS, Kopecky SL, Chaitman BR, et al. Idiopathic short QT interval: a new clinical syndrome? Cardiology. 2000;94:99-102.
2. Gussak I, Bjerregaard P. Short QT syndrome: 5 years of progress. J Electrocardiol. 2005;38:375-7.
3. Algra A, Tijssen JGP, Roelandt JRTC, Pool J, Lubsen J. QT interval variables from 24-Hour electrocardiography and the 2-year risk of sudden death. Br Heart J. 1993;70:43-8.
4. Viskin S, Zeltsner D, Ish-Shalom M, Katz A, Glikson M, Justo D, et al. Is idiopathic ventricular fibrillation a short QT syndrome? Comparison of QT intervals of patients with idiopathic ventricular fibrillation and healthy controls. Heart Rhythm. 2004;1:587-91.
5. Fei L, Camm AJ. Shortening of the QT interval immediately preceding the onset of idiopathic spontaneous ventricular tachycardia. Am Heart J. 1995;130:915-7.
6. Kontny F, Dale J. Self-terminating idiopathic ventricular fibrillation presenting as syncope: a 40-year follow-up report. J Intern Med. 1990;227:211-3.
7.
8. Anttonen O, Junttila MJ, Rissanen H, Reunanen A, Viitasalo M, Hiukuri HV, et al. Prevalence and prognostic significance of short QT interval in a middle-aged Finnish population. Circulation. 2007;116:714-20.
9. Gallagher MM, Magliano G, Yap YG, Padula M, Morgia V, Postorino C, et al. Distribution and prognostic significance of QT intervals in the lowest half centile in 12,012 apparently healthy persons. Am J Cardiol. 2006;98:933-5.
10. Brugada R, Hong K, Dumaine R, Cordeiro JM, Gaita F, Borggrefe M, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109:30-5.
11. Bellocq C, Van Ginnneken CA, Bezzina CR, Alders M, Escande D, Mannens MM, et al. Mutation in the KCNQ1 gene leading to the short QT- interval syndrome. Circulation. 2004;109:2394-7.
12. Hong K, Piper DR, Diaz-Valdecantos, Brugada J, Oliva A, Burashnikov E, et al. De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero. Cardiovasc Res. 2005;68:433-40.
13. Priori SG, Pandit SV, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, et al. A novel form of short QT syndrome (SQT3) is cause by mutation in the HCNJ2 gene. Circ Res. 2005;96:800-7.
14. Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442-9.
15. Anttonen O, Vaananen H, Junttila J, Huikuri HV, Viitasalo M. Electrocardiographic transmural dispersion of repolarization in patients with inherited short QT syndrome. Ann Noninvasive Electrocardiol. 2008;13:295-300.
16. Patel C, Antzelevitch C. Cellular basis for arrhythmogenesis in an experimental model of the SQT1 form of the short QT syndrome. Heart Rhythm. 2008;5:585-90.
17. Gupta P, Patel C, Patel H, Narayanaswamy S, Malhotra B, Green JT, et al. Tp-e/QT ratio as an index of arrhythmogenesis. J Electrocardiol. 2008;41:567-74.
18. Anttonen O, Junttila MJ, Maury P, Schimpf R, Wolpert C, Borggrefe M, et al. Differences in twelve-lead electrocardiogram between symptomatic and asymptomatic subjects with short QT interval. Heart Rhythm. 2009;6:26771.
19. Schimpf R, Bauersfeld U, Gaita F, Wolpert C. Short QT syndrome: successful prevention of sudden cardiac death in an adolescent by implantable cardioverter-defibrillator treatment for primary prophylaxis. Heart Rhythm. 2005;2:4167.
20. Patel Ch, Yan GX, Antzelevitch C. Short QT Syndrome: From bench to bedside. Circ Arrhythm Electrophysiol. 2010;3 401-8.
21. Schimpf R, Antzelevitch C, Haghi D, Giustetto C, Pizzuti A, Gaita F, et al. Electromechanical coupling in patients with the short QT syndrome: further insights into the mechanoelectrical hypothesis of the U wave. Heart Rhythm. 2008;5:2415.
22. Sugishita Y, Iida K, O'Rourke MF, Kelly R, Avolio A, Butcher D, et al. Echocardiographic and electrocardiographic study of the normal kangaroo heart. Aust N Z J Med. 1990;20:1605.
23. Giata F, Giustetto C, Bianchi F, Wolpert C, Schimpf R, Riccardi R, et al. Short QT syndrome: a familial cause of sudden death. Circulation. 2003;108:965-70.
24. Wolpert C, Schimpf R, Veltmann C, Giustetto C, Gaita F, Borggrefe M. Clinical caracteristics and treatment of short QT syndrome. Expert Rev Ther. 2005;4:611-7.
25. Giustetto C, Di Monte F, Wolpert C, Borggrefe M, Schimpf, Sbragia P, et al. Short QT syndrome: clinical findings and diagnostic-therapeutic implications. Eur Heart J. 2006;27:24407.
26. Wolpert C, Schimpf R, Giustetto C, Antzelevitch C, Cordeiro JM, Dumaine R, et al. Further insights into the effect of quinidine in short QT syndrome caused by a mutation in HERG. J Cardiovasc Electrophysiol. 2005;16:548.
27. Watanabe H, Makiyama T, Koyama T, Kannankeril PJ, Seto S, Okamura K, et al. High prevalence of early repolarization in short QT syndrome. Heart Rhythm. 2010;7:64752.
28. Bazett HC. An analysis of the time-relations of electrocardiograms. Heart J. 1920;7:35370.
29. Kobza R, Roos M, Niggli B, Abacherli R, Lupi GA, Frey F, et al. Prevalence of long and short QT in a young population of 41,767 predominantly male Swiss conscripts. Heart Rhythm. 2009;6:6527.
30. Rautaharju PM, Zhou SH, Wong S, Calhoun HP, Berenson GS, Prineas R, et al. Sex differences in the evolution of the electrocardiographic QT interval with age. Can J Cardiol. 1992;8:6905.
31. Gollob MH, Redpath CJ, Roberts JD. The short QT syndrome proposed diagnostic criteria. J Am Coll Cardiol. 2011;57:802-12.
32. Schulze-Bahr E, Breithardt G. Short QT interval and short QT syndromes. J Cardiovasc Electrophysiol. 2005;16:397-8.
33. Bjerregaard P, Gussak I. Short QT syndrome: mechanisms, diagnosis and treatment. Nat Clin Pract Cardiovasc Med. 2005;2:847.
34. Schimpf R, Wolpert C, Bianchi F, Giustetto C, Gaita F, Bauersfeld U, et al. Congenital short QT syndrome and implantable cardio- verter defibrillator treatment: inherent risk for inappropriate shock delivery. J Cardiovasc Electrophysiol. 2003;14:12737.
35. Gaita F, Giustetto C, Bianchi F, Schimpf R, Haissaguerre M, Calo L, et al. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol. 2004;43:14949.
36. Schimpf R, Antzelevitch C, Hsu LF, Schickel S, Pollevick GD, Cordeiro JM, et al. The QT-intervalin patients with a Brugada syndrome: is a shortening of the QT-time an existing and relevant ECG-pattern. Heart Rhythm. 2007;4:S188.
37. Lu LX, Zhou W, Zhang X,
38. Hong K, Bjerregaard P, Gussak I, Brugada. Short QT syndrome and atrial fibrillation caused by mutation in KCNH2. J Cardiovasc Electrophysiol. 2005;16:3946.
Recibido: 30 de marzo del 2012.
Aprobado: 1 de junio del 2012.
Dra. Marleny Cruz Cardentey. Hospital Clinicoquirúrgico "Hermanos Ameijeiras".

{kind=link}