










 Servicios personalizados
Servicios personalizados Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Las pasturas muestran caracteres modificables y heredables en el tiempo, además de una gran variación en hábitos de crecimiento y sistemas de reproducción. Son importantes como especies forrajeras, además de ser rápidas colonizadoras de ambientes degradados, y tener alto potencial ornamental como cespitosas (Capstaff y Miller, 2018). En estas especies, pertenecientes a la familia Poaceae, se observa una amplia gama de comportamientos reproductivos (autopolinización, polinización cruzada, apomixis y propagación vegetativa) (Acuña et al., 2019). La complejidad y variación en los sistemas reproductivos suele ir acompañada de alto grado de plasticidad, lo que indica niveles significativos de interacción genotipo-ambiente para los caracteres agronómicos (Peltier et al., 2018).
Tradicionalmente, el mejoramiento genético de pasturas se basa en técnicas convencionales, dependientes de la variabilidad que ocurre naturalmente en ecotipos adaptados, poblaciones naturalizadas y cultivares viejos. El mejoramiento genético convencional, basado en rasgos morfo fisiológicos, es laborioso y requiere experimentos repetidos en múltiples entornos (Collard y Mackill, 2008). Es por ello que las herramientas moleculares pueden acelerar y mejorar sustancialmente los programas de mejora.
Desde el descubrimiento del ADN como portador de la información genética de la célula, los científicos se han dedicado constantemente a desarrollar herramientas que permitan manipular y modificar el genoma. El desarrollo de diversas técnicas moleculares permitió un avance en el mejoramiento genético de los cultivos de interés agrícola y la introducción de nuevos enfoques, para acortar los ciclos reproductivos de las plantas. Se ha demostrado que las técnicas novedosas desarrolladas en los últimos 20 años, como la selección genómica y el fenotipado de alto rendimiento (HTP, por sus siglas en inglés) aceleran el fitomejoramiento (Ahmar et al., 2020).
La ingeniería genética y los métodos moleculares también desempeñan una función importante para desarrollar cultivos con características deseables mediante la transformación genética, la mutagénesis, la fusión de protoplastos, el cultivo in vitro, el rescate de embriones, entre otros (Watson et al., 2018). Estudios recientes proponen técnicas, como la secuenciación genómica a gran escala y marcadores moleculares de alto rendimiento, para mejorar la reproducción de cultivos de importancia comercial (Mujjassim et al., 2019).
A partir de los antecedentes descritos, el objetivo de esta revisión es fundamentar la importancia de la aplicación de herramientas moleculares en los programas de mejoramiento genético de pasturas.
Uso de métodos moleculares para evaluar la diversidad genética en pasturas. La caracterización de la diversidad genética de una población es necesaria para un mejor uso de los recursos genéticos en los programas de mejoramiento y conservación de la biodiversidad. Por estas razones, el conocimiento de la diversidad genética del germoplasma disponible es esencial en la selección de materiales para el cultivo o progenitores para el desarrollo de cultivares (Kuwi et al., 2018). Para la evaluación de la diversidad genética se pueden utilizar diferentes herramientas, entre las que se destacan los marcadores moleculares. Estos se desarrollan mediante el análisis de fragmentos, matrices de hibridación o métodos basados en secuenciación del ADN (Loera-Sánchez et al., 2019).
Los marcadores moleculares son segmentos de ADN, que están asociados con una parte del genoma y que se pueden identificar mediante un ensayo simple (Nadeem et al., 2018). Se basan en diferencias en la secuencia de ADN, por lo que no están sujetos a la influencia ambiental (Hamouda, 2019). Son abundantes en todo el genoma y los ensayos correspondientes se pueden realizar en cualquier momento durante el desarrollo de la planta (Nadeem et al., 2018).
Métodos basados en análisis de fragmentos. Los métodos basados en ADN se clasifican en técnicas de análisis de fragmentos (marcadores), matrices de hibridación (detectan polimorfismos de ADN mediante hibridación de una muestra de ADN y un matriz marcada con sondas) y métodos basados en secuenciación que detectan polimorfismos de ADN por secuenciación (Loera-Sánchez et al., 2019).
Entre las metodologías que se basan en análisis de fragmentos, que detectan los polimorfismos de ADN mediante la comparación del tamaño de las secuencias, se encuentran el polimorfismo de longitud de fragmento amplificado (AFLP), el ADN polimórfico amplificado aleatoriamente (RAPD), sitios de unión intercebador (iPBS), polimorfismos amplificados relacionados con la secuencia (SRAP), repeticiones de secuencia intersimple (ISSR), polimorfismos de longitud de fragmentos de restricción (RFLP) y repeticiones de secuencia simple o microsatélites (SSR).
Los marcadores moleculares pueden ser dominantes o codominantes, en dependencia de los alelos que puedan identificar. Los marcadores dominantes (AFLP, RAPD, iPBS, SRAP e ISSR) no son capaces de identificar los individuos heterocigotos, lo que constituye una limitación, pero tienen como ventaja que se pueden producir a bajo costo, sin que sea necesaria información sobre la secuencia de las especies objetivo. Esto los convierte en los sistemas de elección para muchas especies de pastizales que no han sido secuenciadas (Loera-Sánchez et al., 2019).
Los sistemas de marcadores codominantes, como el SSR, permiten la estimación de la diversidad genética y se basan en frecuencias alélicas. El desarrollo de estos marcadores requiere información de secuenciación a priori, lo que constituye una limitación en el caso de genomas que no han sido secuenciados. Sin embargo, una vez obtenidos, los conjuntos de cebadores SSR se pueden estandarizar fácilmente y utilizar por múltiples laboratorios (Loera-Sánchez et al., 2019; Romero et al., 2019). Por estas razones, se consideran marcadores ideales en estudios de diversidad genética, debido a la facilidad de su aplicación, alta reproducibilidad, análisis rápido, bajo costo y mayor diversidad alélica (Tibihika et al., 2019).
Los marcadores moleculares se usan ampliamente en la identificación de cultivares y especies, en el establecimiento de relaciones evolutivas entre diferentes grupos de plantas, en la evaluación de la variabilidad genética entre poblaciones, mapeo genético y selección asistida. Por estas razones, se consideran herramientas básicas y útiles en programas de mejora genética (Carrodeguas-Gonzalez y Zuñiga-Orozco, 2020).
Con el propósito de evaluar la diversidad genética en gramíneas, varios autores han utilizado diferentes marcadores moleculares que se muestran en la figura 1.

Figura 1 Estudios relevantes de diversidad genética a partir del uso de marcadores moleculares en diferentes gramíneas.
En los últimos años, ha crecido el interés por la evaluación de la diversidad genética en poblaciones de pasturas mediante el uso de marcadores moleculares, ya que este tipo de estudio constituye el requisito previo para el éxito en cualquier programa de selección (Carrodeguas-Gonzalez y Zúñiga-Orozco, 2020). Recientemente, Luo et al. (2020) mediante el uso de marcadores morfológicos (SNPs, SRAP e ISSR) analizaron la diversidad genética en 49 accesiones de Stenotaphrum secundatum (Walt.) Kuntze, un césped popular en jardines de regiones tropicales y subtropicales, con el objetivo de comparar la eficiencia de dichas técnicas. Estos autores demostraron que en la especie analizada los tres marcadores moleculares utilizados son muy efectivos para este tipo de estudio.
Matrices de hibridación. Las matrices de hibridación de ADN son técnicas de alto rendimiento, basadas en la hibridación de secuencias de ADN con una matriz de sondas marcadas, que están unidas a una superficie sólida. Después de eliminar el ADN no hibridado, las hibridaciones exitosas se visualizan mediante fluorescencia o quimioluminiscencia (Loera-Sánchez et al., 2019). Los métodos basados en matrices de hibridación, como las matrices SNP y la tecnología matriz de diversidad (DArT), se han utilizado para medir la variación genética de pastizales (Blackmore et al., 2015).
La tecnología basada en matrices de diversidad (DArT) utiliza representaciones genómicas de las poblaciones objeto de análisis. Dichas representaciones se generan mediante cortes de ADN de múltiples plantas con enzimas de restricción. Luego se enriquecen ciertos fragmentos con el uso de cebadores selectivos y, finalmente, los fragmentos se clonan en una biblioteca de plásmidos. A diferencia de las matrices SNP, esta tecnología de matrices no requiere información de secuenciación a priori, lo que reduce en gran medida sus costos de desarrollo (Loera-Sánchez et al., 2019).
Los polimorfismos de un solo nucleótido (SNP) son mutaciones codominantes heredadas, que ocurren en el nivel de base única en regiones del genoma, codificantes o no codificantes. Los SNP se identifican comparando las secuencias de múltiples plantas de una población y se pueden utilizar para estudiar la diversidad genética (Nybom et al., 2014). En los próximos años, con la reducción del costo de la secuenciación, se espera que estos marcadores sean los más utilizados e idóneos para evaluar variabilidad en pasturas.
Métodos basados en secuenciación. La secuenciación de nueva generación (NGS, por sus siglas en inglés) agrupa un conjunto de tecnologías, diseñadas para secuenciar gran cantidad de segmentos de ADN de forma masiva y en paralelo, en menor cantidad de tiempo y a menor costo por base. Inicialmente se utilizó para detectar variantes de nucleótido único, y se ha desarrollado para otro tipo de variantes, como inserciones, deleciones y grandes reordenamientos (Raza y Shahi, 2020).
NGS brinda la oportunidad de explorar la diversidad genética en pasturas y sus parientes silvestres, a una escala mucho mayor de lo que era posible con tecnologías anteriores. El análisis del acervo genético primario y de los parientes silvestres más distantes tiene el potencial de identificar genes y alelos que se pueden usar para mejorar el rendimiento en los pastos.
Marcadores moleculares para la identificación de plantas apomícticas. Durante la reproducción sexual de las angiospermas, el polen es indispensable para la fertilización y para asegurar la formación de semillas sexuales. Sin embargo, en muchas plantas conocidas como apomícticas, la formación de la semilla ocurre sin necesidad de la doble fertilización característica de las angiospermas.
La apomixis es una forma de reproducción asexual mediante semillas, que origina plantas genéticamente idénticas a la planta madre, o sea, constituye un método de clonación natural. Estas semillas se forman a partir de tejidos maternos del óvulo, y evitan los procesos de meiosis y fertilización. Este fenómeno está relacionado estrechamente con el nivel de ploidía, de forma que los genotipos diploides presentan, generalmente, reproducción sexual, mientras que los poliploides son apomícticos (Soliman et al., 2019). La apomixis puede ser gametofítica o esporofítica. En la esporofítica (embrionía adventicia), el embrión se desarrolla directamente en el óvulo a partir de una célula somática (generalmente de la nucela o tegumento), fuera del saco embrionario sexual. Este tipo de apomixis se ha descrito en los géneros Poa, Oryza y Paspalum (Fiaz et al., 2021). En la gametofítica, el saco embrionario se obtiene vía aposporia o diplosporia. El embrión se forma luego por embriogénesis, independientemente de la fertilización (partenogénesis), y el endospermo se desarrolla de forma autónoma o después de la fertilización de los núcleos polares (pseudogamia) (Henderson et al., 2017).
La apomixis está presente en más de 400 especies vegetales, que representan, aproximadamente, 40 familias. Se ha informado la aparición de embriones adventicios en 148 géneros, aposporia en 110 y diplosporia en 68 géneros (Hojsgaard et al., 2014).
La diplosporia consiste en la mitosis que experimentan las células madre de las megásporas para formar un saco embrionario, no reducido (no ocurre meiosis, por lo que no se reduce el número cromosómico). Las células de iniciación apomíctica se originan a partir de la célula madre de las megásporas y, finalmente, se convierten en embriones. Este tipo de apomixis se ha observado en la familia Poaceae, en los géneros Paspalum, Tripcacum, Eragrostis y Elymus (Quero-Carrillo et al., 2010).
Durante la aposporia, las células somáticas que se encuentran cerca de las células madre de la megáspora son las que forman un saco embrionario no reducido. Estas células se someten a tres rondas de mitosis, y finalmente se convierten en embriones (Schmidt, 2020). Se han registrado en la familia Poaceae, en los géneros Megathyrsus, Paspalum, Brachiaria, Bouteloua, Cenchrus y Pennisetum (Quero-Carrillo et al., 2010).
La figura 2 muestra los tipos de apomixis.
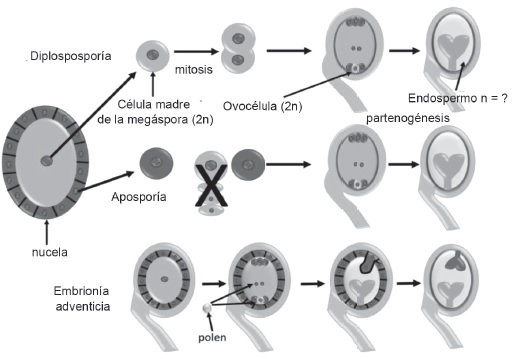
Figura 2. Tipos de apomixis en angiospermas. Primero se observa diplosporia, presente en poaceas, donde las células madre de las megásporas experimentan mitosis para formar un saco embrionario no reducido. En la aposporia, también presente en poáceas, el saco se forma a partir de células de la nucela. Por último, se observa embrionía adventicia.
En la agricultura, la apomixis constituye una ventaja para mantener genotipos superiores de diferentes cultivos. La perspectiva de clonar genotipos híbridos con características de interés agronómico puede representar una ayuda importante para los productores agrícolas de los países en desarrollo. Esto les permitiría sostener rendimientos altos, al usar parte de las semillas cosechadas sin pérdidas en la producción, debido a la segregación de caracteres y la depresión por endogamia.
Entre otras ventajas, la expresión de la apomixis reduce al mínimo el aislamiento físico requerido para preservar líneas genéticas homocigotas. Los nuevos híbridos interespecíficos e intergenéricos se pueden obtener y propagar fácilmente, lo que permite el desarrollo de genotipos mejor adaptados a los distintos ambientes.
En un inicio, en los programas de mejora genética en pasturas, la generación de híbridos entre especies apomícticas y no apomícticas se obstaculizó por las diferencias de ploidía. Este fenómeno constituyó un problema para los mejoradores hasta que el avance de la tecnología permitió la creación de biotipos poliploidizados, de interés para su cruce con los apomícticos.
La generación de híbridos en especies apomícticas incluye los procedimientos siguientes, según describe Quero-Carrillo et al. (2010):
Los individuos sexuales diploides de la especie de interés agronómico se poliploidizan en laboratorio mediante cualquier agente inhibidor de la formación del uso acromático durante la mitosis, lo que induce la sexualidad poliploide.
Los poliploides apomícticos se utilizan como polinizadores y la fertilización no representa un problema, cuando se usan individuos con el mismo nivel de ploidía con respecto a la planta hembra.
La segregación de individuos apomícticos y sexuales mantiene la proporción mendeliana 1:1 en la descendencia. Los individuos sexuales, genéticamente recombinados, se pueden integrar al grupo de progenitores futuros.
Los híbridos apomícticos se evalúan para los atributos de interés, dado que el vigor híbrido queda integrado en el genoma.
A partir de este esquema, la identificación precoz del modo reproductivo en poblaciones híbridas ayudaría a identificar genotipos apomícticos con atributos de interés para que se conviertan en nuevos cultivares, capaces de mantener los caracteres deseados.
Tradicionalmente, en pasturas se utilizan descriptores morfológicos, así como el análisis de sacos embrionarios al microscopio para diferenciar genotipos sexuales de apomícticos (Savidan, 2000). A partir de 1990, con el desarrollo de técnicas biotecnológicas se comienzan a utilizar marcadores moleculares, con la ventaja de que brindan resultados en poco tiempo; además de que se puede utilizar cualquier tejido vegetal de plantas, sin importar el estado de crecimiento (Poblete-Vargas et al., 2018). Se han realizado diversos estudios en busca de marcadores moleculares asociados a la apomixis en pastos. La tabla 1 muestra los más relevantes.
Tabla 1 Marcadores moleculares utilizados para la identificación de plantas apomícticas o sexuales en poáceas.
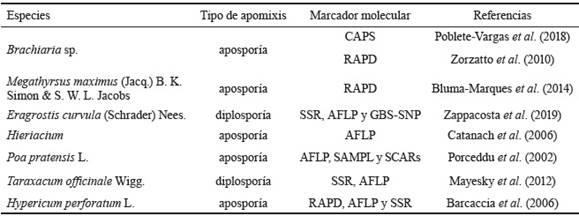
En la actualidad, los polimorfismos de un solo nucleótido (SNP) son los marcadores moleculares que más se utilizan en estudios genéticos, y como apoyo a ellos surge en los últimos años el genotipado por secuenciación (GBS) (Zappacosta et al., 2019). Los primeros mapas de ligamiento fueron desarrollados por Worthington et al. (2016) para una especie de pasto apomíctica poliploide (Brachiaria decumbens Stapf.) mediante la utilización de marcadores SNP, generados por GBS. Este mapa de ligamiento se ha utilizado para identificar marcadores vinculados a la región genómica específica de aposporia (ASGR).
En un estudio realizado por Zappacosta et al. (2019) se construyó el primer mapa de ligamiento saturado de Eragrostis curvula, donde se utilizaron marcadores moleculares tradicionales (AFLP y SSR) y de alto rendimiento (GBS-SNP). Estos autores identificaron el locus que controla la diplosporia y regiones reguladoras putativas que afectan la expresividad de dicho rasgo. Los análisis de locus de rasgo cuantitativo (QTL, por sus siglas en inglés), que tienen que ver con la expresividad de diplosporia en los híbridos F1, entre una variedad sexual y otra apomíctica, revelaron la presencia de dos QTL principales, ubicados a 3,27 y 15 cM del locus de diplosporia.
El análisis molecular de poblaciones híbridas F1, provenientes de individuos homocigotos, para la apomixis permite la construcción de mapas genéticos y la localización de marcadores moleculares asociados con la apomixis. Estas poblaciones están compuestas por individuos genéticamente cercanos, pero que tienen diferentes modos de reproducción, lo que permite hacer estudios de expresión para la identificación de genes candidatos que pudieran regular la apomixis.
Se han realizado muchos esfuerzos para transferir la apomixis a otros cultivos vía transformación genética, pero no se han alcanzado buenos resultados. Para aplicar la transgénesis es importante conocer las vías moleculares y los genes implicados en la apomixis. Con este objetivo, se desarrollaron varios estudios basados en hibridaciones interespecíficas entre plantas sexuales y apomícticas: el análisis del proceso en apomícticas naturales y la identificación de mutantes de especies sexuales que imitan la apomixis (Garbus et al., 2017; Selva et al., 2017).
El proceso de desarrollo de semillas apomícticas es complejo y comprende tres componentes: apomeiosis (que conduce a la formación de óvulos no reducidos), partenogénesis (desarrollo de embriones sin fertilización) y desarrollo funcional del endospermo (Kaushal et al., 2019). Los componentes de apomeiosis y partenogénesis de la apomixis en otras gramíneas se heredan, generalmente, como un solo locus dominante, conocido como región genómica específica de aposporia (ASGR), identificada en Pennisetum (Ozias-Akins y Van Dijk, 2007). La ASGR contiene segmentos ricos y pobres en genes, donde varios genes pueden desempeñar una función determinada en el desarrollo apomíctico, así como muchas clases de elementos transponibles (Fiaz et al., 2021).
Conne et al. (2015) encontraron en Cenchrus y Pennisetum un gen candidato para la partenogénesis: ASGR-BABY BOOM (ASGR-BBML). En estudios posteriores, este gen se combinó con otros, y se produjo una metodología para la obtención de semillas en arroz por vía asexual (Khanday et al., 2019; Wang et al., 2019).
Aplicaciones de CRISPR en la mejora genética de pasturas. Durante el año 1987, investigadores de la Universidad de Osaka, en Japón, descubrieron en el genoma de Echerichia coli cinco repeticiones de 29 nucleótidos, espaciados por 32 nucleótidos. Posteriormente, se identificaron en otras bacterias, como Haloferax mediterranei, Streptococcus pyogenes, Anabaena sp. PCC 7120 y Mycobacterium tuberculosis. El término CRISPR (repeticiones palindrómicas cortas agrupadas y regularmente espaciadas, por sus siglas en inglés) se designó para hacer referencia a estas secuencias repetidas. Se identificaron, además, genes asociados a CRISPR, denominados Cas, los que codifican para endonucleasas de restricción, conocidas como caspasas (Concepción-Hernández, 2018).
Se han identificado tres sistemas principales CRISPR, que tienen en común la presencia del componente CRISPR, un ARN guía y el componente Cas, que difiere para el tipo de sistema. En CRISPR I se activa Cas3, en el segundo sistema Cas9 y en CRISPR III Cas6 (Liu et al., 2020).
El segundo sistema, CRISPR-Cas9, tiene dos componentes: el primero es una enzima (Cas9), que funciona como unas tijeras que cortan el ADN (las bacterias la utilizan para desarmar el genoma de virus invasores). El otro componente (CRISPR) es un ARN, que guía a las tijeras hacia una secuencia de nucleótidos específica, conocida como objetivo o diana. Una vez encontrado el objetivo o target, la Cas9 abre la cadena de ADN, y posibilita la modificación de la secuencia de nucleótidos (Zúñiga-Orozco, 2018). La figura 3 muestra el modo de acción del complejo CRISPR-Cas9.

Figura 3. Tecnología de edición genómica CRISPR/Cas9. La polimerasa junto al ARN guía abre la molécula de ADN en un punto específico o target, el cual se vuelve a cerrar por recombinación homóloga/no homóloga.
El sistema CRISPR / Cas9 de tipo II realiza cortes de doble hebra (DSB), justo antes de un motivo adyacente protoespaciador (PAM) de tres nucleótidos de longitud (NGG). Los DSB se pueden reparar mediante la vía de unión terminal no homóloga propensa a errores (NHEJ, por sus siglas en inglés) o por la vía de reparación de recombinación homóloga (HDR, por sus siglas en inglés), las que pueden generar mutantes (Svitashev et al., 2016). Debido a que CRISPR/Cas9 funciona en trans, se puede crear una mutación en un locus distante del sitio de inserción del transgén. Mediante el mejoramiento tradicional, el transgén se puede eliminar sin afectar la mutación (Xu et al., 2016). Estos mutantes son muy diferentes de las plantas transgénicas tradicionales, y pueden requerir menos o ninguna supervisión regulatoria (Liu et al., 2018).
En muchas especies de pastos, los estudios genéticos se dificultan debido a la alta autoincompatibilidad y a la condición poliploide, como por ejemplo Megathyrsus virgatum L. (Martínez-Reyna y Vogel, 2002). Para superar estas limitaciones, Liu et al. (2018) exploraron la viabilidad de utilizar CRISPR/Cas9 para la mutagénesis dirigida en un cultivar tetraploide de M. virgatum. Para ello establecieron un ensayo transitorio mediante el uso de protoplastos de mesófilo, con el objetivo de validar la actividad CRISPR/Cas9. Estos autores demostraron además, que esta metodología es eficaz para crear mutaciones dirigidas de modo simultáneo.
CRISPR/Cas9 se ha utilizado ampliamente para la mutagénesis específica en un gran número de especies de plantas herbáceas de importancia agronómica, que incluyen arroz, sorgo, maíz y trigo (Svitashev et al., 2016). Para los pastos, Capstaff y Miller (2018) hacen referencia a genes candidatos relacionados con procesos fisiológicos importantes para la planta. Este es el caso de los genes asociados a la familia del superóxido dismutasa férrica, responsables de la acumulación de biomasa, así como de los genes que se identifican en el enanismo, que se pueden utilizar para reducir el tamaño en especies de porte alto.
En especies forrajeras que no son gramíneas, como Medicago sativa L., se identificaron genes candidatos para mejorar caracteres de interés económico. A partir de ellos se podría utilizar el programa Biomercator para analizar genes ortólogos en poáceas bien estudiadas, como el arroz, y aumentar así la posibilidad de encontrar estos genes en gramíneas que se usan como pasturas. En M. sativa se encontraron genes como CONSTANS-LIKE, asociados a la floración y a la altura del tallo floral, lo que puede ser importante para especies de porte alto. Li et al. (2017) hacen referencia a genes como Arabidopsis Enhanced Drought Tolerance1, HSP23 y a genes de la familia MsHSP, que mejoran la acumulación de biomasa, el contenido de sacarosa y de clorofila bajo estrés hídrico. De manera similar, se ha registrado que los genes MsERF9 y MsERF11 confieren tolerancia a la salinidad (Chen et al., 2012).
CONCLUSIONES
Los marcadores moleculares permiten estudios de diversidad genética en pasturas. Con el avance de la tecnología, los marcadores más utilizados serán los SNP.
La apomixis es una forma de reproducción en muchas especies de pastos y constituye una herramienta necesaria para mantener y reproducir híbridos con características de interés agronómico. Por medio de marcadores moleculares es posible reconocer a edades tempranas las plantas apomícticas.
Con tecnologías de edición genómica, como CRISPR, se abre la posibilidad de mejorar caracteres en pasturas. Los caracteres candidatos son los relacionados con: mayor tolerancia a estrés biótico y abiótico (especialmente sequía), aceleración de la tasa de rebrote, mayor cantidad y rapidez de acumulación de biomasa y contenido de proteína para forrajes.

