










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Ortopedia y Traumatología
versión On-line ISSN 1561-3100
Rev Cubana Ortop Traumatol v.19 n.2 Ciudad de la Habana jul.-dic. 2005
¨Frank País¨ Ciudad de La Habana, Cuba
Enfermedad de McCune-Albright
Dr. Luis Oscar Marrero Riverón,1 Dra. Vilma Rondón García,2 Dra. Martha Melo Víctores,3 Dra. Lina Aurora Chao Carrasco,4 Dra. Hilda Elena Roché Egües 5 y Dr. Jorge Luis Roche Sánchez 6
Marrero Riverón LO, Rondón García V, Melo Víctores M, Chao Carrasco LA, Roché Egües HE y Roche Sánchez JL. Diagnóstico y seguimiento evolutivo de un paciente con Enfermedad de McCune-Albright. Rev Cubana Ortop Traumatol. 2005;19(2)
Resumen
Se aborda el diagnóstico y seguimiento de la Enfermedad de McCune-Albright en un paciente masculino de 8 años de edad, afectado de esta rara entidad.
Palabras clave: Enfermedad de McCune-Albright, diagnóstico, exámenes complementarios, imágenes por radionúclidos, cintigrafía ósea.
Las displasias fibrosas constituyen un grupo de displasias en las cuales existe un fallo en la formación de hueso esponjoso.1 La Enfermedad de McCune-Albright es un síndrome de rara aparición caracterizado por una displasia fibrosa poliostótica, manchas café-au-lait y varias hiperfunciones endocrinas que conducen comúnmente a una pubertad precoz.2-4 En ocasiones puede acompañarse de otros tipos de alteraciones.4-6 En el 95 % de los casos reportados, el paciente es del sexo femenino.2
El objetivo del presente trabajo es analizar el diagnóstico y seguimiento de un paciente con la afección de referencia.
Presentación del caso
Paciente masculino de 8 años de edad, de piel blanca, procedente de la provincia Pinar del Río, que acudió en enero del 2002 al CCOI "Frank País" por presentar claudicación en la marcha y dolor en la cadera izquierda, con aproximadamente 6 meses de evolución.
Examen físico
Paciente normolíneo que claudica en la marcha a expensas del miembro inferior izquierdo.
Talla: 106 cm
Peso: 22 kg
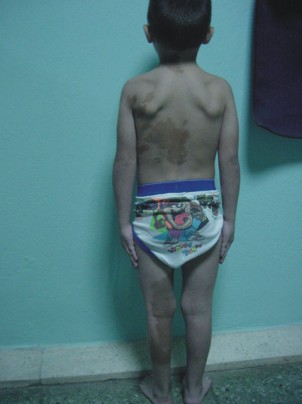
Manchas café-au-lait distribuidas en cara, tronco, espalda, glúteos, genitales externos y muslos (fig. 1).
Genitales externos en estadio I de Tanner.
Limitación dolorosa en la flexión, abducción y rotación externa de ambas caderas. Tono muscular disminuido, atrofia muscular.
Soplo sistólico IV/VI en válvula mitral. Ausencia de arritmias.
Pulso radial 100/ min .
Hepatomegalia de aproximadamente 2 cm.
Exámenes de laboratorio clínico
Hemoglobina: 13,6 g/l
Hematocrito: 0,40
Eritrosedimentación: 8 mm/h
Calcio: 2,3 mmol/l
Fósforo: 1,7 mmol/l
Fosfatasa alcalina: 206 ?/l
TGP:17,4
TGO: 10,0
Serología: no reactiva
HIV: negativo
Proteína C reactiva: negativa
Factor reumatoideo: negativo
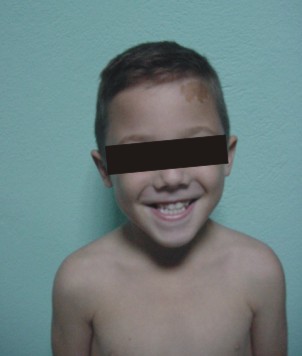
FIG. 1. A) Manchas café-au-lait en región frontal izquierda. B) Distribución de las manchas café-au-lait en el dorso.
Estudios imagenológicos
- Survey óseo radiográfico (fig. 2)
- Pelvis ósea: coxa vara bilateral con aumento del diámetro transversal de los 2/3 superiores de ambos fémures, cortical afinada y cavidad medular con aspecto de "vidrio esmerilado". Incurvación en varo del eje longitudinal del fémur.
- Manos y pies: expansión de las diáfisis de los metacarpianos y metatarsianos, corticales afinadas y rarefacción ósea de las cavidades medulares por alteraciones de la textura ósea.
- Tibias y peronés: ensanchamiento del diámetro transversal en el tercio medio del peroné derecho y de toda la diáfisis del peroné izquierdo en cuya mitad superior, la cortical está afinada y centralmente la cavidad medular tiene aspecto de "vidrio esmerilado".
- Húmero, cúbito y radio izquierdos: con lesiones diafisarias similares a las descritas.
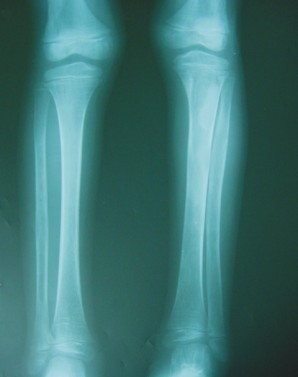

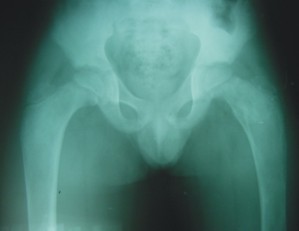
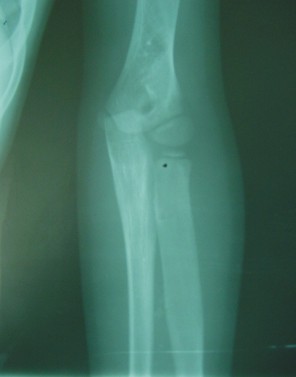
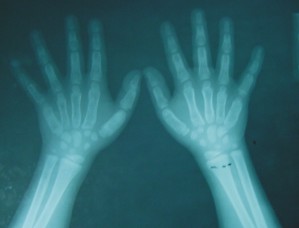
FIG. 2. A) Radiografía simple de ambas tibias y peronés. B) Radiografía simple de ambos pies. C) Radiografía simple de la pelvis. Coxa vara bilateral. D) Radiografía simple del codo izquierdo.E) Radiografía simple de ambas manos.
- Pelvis ósea: coxa vara bilateral con aumento del diámetro transversal de los 2/3 superiores de ambos fémures, cortical afinada y cavidad medular con aspecto de "vidrio esmerilado". Incurvación en varo del eje longitudinal del fémur.
Se observó expansión de la porción esponjosa y alteraciones de la textura ósea como hallazgos radiográficos frecuentes en los huesos tubulares afectados.
- Tomografía en espiral del cráneo
Expansión del díploe craneal, tanto de la cortical externa como de la interna, con moteado donde predomina la esclerosis (fig. 3 A). Los agujeros anatómicos están estrechados por el crecimiento óseo endóstico (fig. 3 B). Mayor afección izquierda.
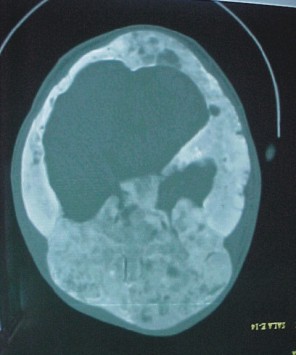
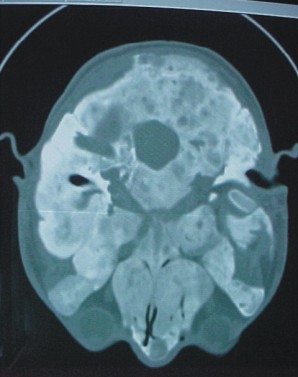
FIG. 3. A y B) Imágenes de tomografía en espiral del cráneo.
- Survey óseo gammagráfico (fig 4)
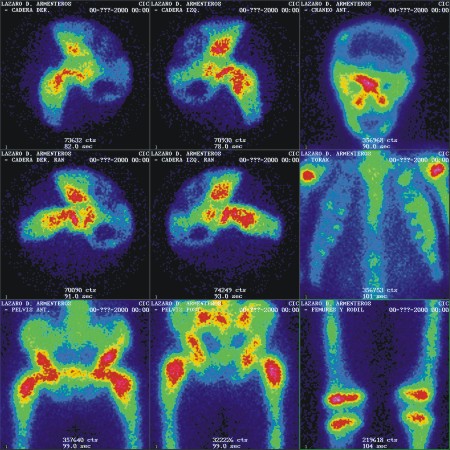
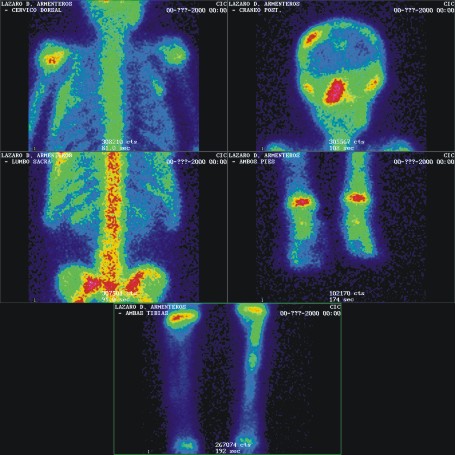
FIG. 4. A y B) Survey óseo gammagráfico.
En las imágenes estáticas mediante el uso del colimador LEHR (Low Energy High Resolution) y 700 kiloconteos, se aprecian múltiples lesiones hipercaptantes, homogéneas en cráneo (región fronto-parietal derecha y macizo facial), parrilla costal izquierda, ambas caderas, diáfisis tibial izquierda, manos y pies).
Una vez establecido el diagnóstico de Enfermedad de McCune-Albright, se realizaron interconsultas con especialistas en Endocrinología y Genética Médica.
Seguimiento evolutivo
El paciente presentó aumento de los niveles séricos de las enzimas hepáticas cuando se aplicó tratamiento con pamidronato, que posteriormente disminuyeron pero no se han normalizado.
En junio del 2002 aumentó la claudicación en la marcha y se detectó en las radiografías fractura de estrés en la región subtrocantérica izquierda, se inmovilizó en espica de yeso por 6 semanas y se logró la consolidación ósea. En julio del 2004 se produjo otra fractura de estrés al nivel de la diáfisis femoral izquierda (fig 5) se decidió intervenir quirúrgicamente para colocar enclavijado intramedular, pero la fosfatasa alcalina sérica se encontraba elevada a 2414, la TGP en 46 mmol/l y la TGO en 51 mmol/l, lo cual contraindicó la intervención y tuvo que inmovilizarse nuevamente.
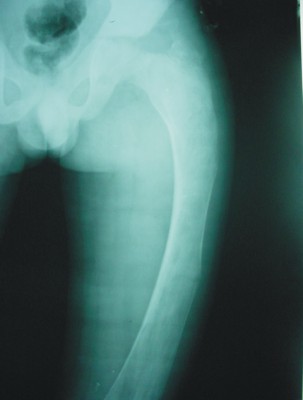
FIG 5. Radiografía simple del fémur izquierdo que muestra fractura patológica
En enero del 2005 existía consolidación ósea, acortamiento de 2 cm y arqueamiento del miembro inferior izquierdo, deambulaba con muletas. Aumentó la estatura en 6 cm (1 por mes) y presentó ganancia de peso de 3 kg. Las cifras de TGP y TGO se mantenían elevadas.
Discusión
La más completa descripción de la entidad de referencia es realizada separadamente por D. McCune y F. Albright en 1937, aunque anteriormente se había reportado casos similares.5
Boston 5 afirma que el síndrome en su forma clásica consiste en al menos 2 de los hallazgos de la triada integrada por la displasia fibrosa poliostótica, las manchas café-au-lait y la hiperfunción autónoma endocrina. Se considera una entidad extremadamente rara, cuya incidencia es desconocida, aunque es más frecuente en el sexo femenino. Se detecta a cualquier edad, sin distinción del color de la piel, cualquier hueso puede estar afectado.2,5-7
La forma de presentación clínica de la Enfermedad de McCune-Albright es muy variable. Las manchas café-au-lait se detectan al nacer, el sangramiento vaginal se describe desde la infancia temprana y las manifestaciones óseas pueden ser de curso silente y sólo detectarse cuando comienza la claudicación o por una radiografía incidental. Las alteraciones en los huesos, la piel y los ovarios son las más frecuentes; pero tanto los órganos endocrinos como otros pueden afectarse (tiroides, suprarrenales, hipófisis, hígado y corazón). Por ello, no es rara la asociación con endocrinopatías (acromegalia o gigantismo, hipertiroidismo y enfermedad de Cushing) así como con otras enfermedades (hipofosfatemia, hepatopatías crónicas, taquicardia y raramente, muerte súbita, posiblemente por arritmias cardiacas).1,5, 7
Se descubrió en años recientes que el síndrome de McCune-Albright es el resultado de una mutación somática postzigótica en el gen codificador de la proteína G estimulatoria (Gsa). La proteína G está involucrada en la transmisión de señales hormonales intracelularmente. La mutación específica causante de esta afección produce una activación constitutiva de una cascada de señales intracelulares en ausencia de estimulación hormonal, se mantiene activada la subunidad Gsa, es constante la estimulación de la adenilciclasa y se producen persistentemente altos niveles intracelulares de AMP cíclico, lo cual explica la triada clásica de la enfermedad.1,5,7
La displasia fibrosa en el síndrome de McCune-Albright puede incidir en cualquier hueso, pero más comúnmente afecta en el esqueleto axial, las costillas y los huesos del macizo cráneo-facial, y en el esqueleto apendicular, la tibia y el fémur proximal. Generalmente las afectaciones predominan en uno de los hemicuerpos y varían desde áreas pequeñas y asintomáticas - sólo detectables por gammagrafías ósea - hasta lesiones desfiguradoras con frecuentes fracturas patológicas y atrapamientos de nervios periféricos, trastornos de la visión (ceguera y proptosis) y de la audición (sordera y vértigos). Hacia los 10 años de edad, frecuentemente los pacientes presentan deformidades angulares óseas. La deformidad del cuello femoral causa coxa vara progresiva que conduce a la deformidad en cayado de pastor. Las lesiones pueden permanecer estáticas o empeorar, pero nunca mejoran. 1,5-7
Las manchas café-au-lait en el síndrome de referencia son en realidad, grandes máculas melanóticas que sólo muestran hiperpigmentación de la lámina basal, no son visibles en el niño pequeño y el color varía de carmelita claro a oscuro. Su tamaño es variable, pueden tener una distribución segmentaria y con frecuencia predominan en un hemicuerpo (fig. 1 A y B). Las lesiones individuales no suelen pasar la línea media. Sus bordes son irregulares del tipo costas de Maine, a diferencia de las lesiones de la neurofibromatosis que son de bordes regulares del tipo costas de California (en alusión a las costas de los estados norteamericanos de igual nombre).5-7
Las alteraciones hepáticas varían desde la elevación moderada de las transaminasas hasta la ictericia neonatal y las colestasis crónicas.5
La pubertad precoz, el defecto endocrino más frecuente, es el resultado de una función gonadotropina-independiente, autonómica, de ovarios o testículos; más común en el sexo femenino con presencia de ginecomastias y sangramiento vaginal. A causa de la exposición excesiva a los estrógenos se incrementa la velocidad de crecimiento y se adelanta la madurez esquelética, lo que trae como consecuencia una menor estatura final. 5, 7
Debe realizarse diagnóstico diferencial con las siguientes afecciones: 5, 7
- Displasia fibrosa monostótica
- Neurofibromatosis
- Hipertiroidismo
- Síndrome de Cushing
- Terapia con glucocorticoides
- Terapia con hormonas tiroideas
- Gigantismo y acromegalia
- Raquitismo hipofosfatémico
- Pubertad precoz de tipo central
- Tumor funcionante de ovario
- Síndrome de Mazabraud
- Enfermedad de Paget
- Síndrome de Proteus
Entre los estudios complementarios de laboratorio clínico deben incluirse: 5, 7
- Calcio y fósforo séricos
- Fosfatasa alcalina sérica
- N-telopéptidos e hidroxiprolina urinarios
- Osteocalcina sérica
- Enzimas hepáticas
- Hormonas tiroideas
- TSH
- ACTH sérica
- Cortisol urinario
- Hormona del crecimiento
- Estradiol sérico, FSH y LH, testosterona sérica, en caso de pubertad precoz.
Los estudios imagenológicos deben incluir:
- Ultrasonidos de la pelvis, para detectar y medir quistes de ovarios (generalmente grandes y unilaterales) así como de las partes blandas, para detectar tumoraciones.
- Tomografía axial computadorizada de abdomen y de cráneo, para detectar hiperplasia suprarrenal y atrapamiento de nervios intracraneales.
- Survey óseo radiográfico y determinación de la edad ósea.
- Survey óseo gammagráfico, para detectar sitios asintomáticos (que deben ser comprobados por rayos x).
Histológicamente, las zonas afectadas por la displasia fibrosa presentan abundantes células similares a los fibroblastos con mínima matriz extracelular, exceso de células proosteogénicas que maduran en osteoblastos anormales. Existe un patrón óseo desorganizado, similar a una "sopa alfabética" o de "letras chinas", el osteoide es de forma irregular (retorcido) con un estroma fibroso muy celular. 1,5, 6
El tratamiento de la Enfermedad de McCune-Albright requiere un abordaje multidisciplinario de ortopédicos, endocrinólogos, psicólogos y pediatras o internistas según la edad del paciente.
Los pacientes no necesitan una dieta especial. La actividad física sólo se limita cuando existan lesiones óseas en zonas de carga de peso con riesgo de fracturas patológicas; deben evitarse los deportes de contacto, pero sí mantener un programa de ejercicios. En el tratamiento de la displasia fibrosa, los pacientes asintomáticos no requieren un tratamiento específico, sólo observación. La mayoría de las fracturas se tratan en tracción, las del extremo proximal del fémur requieren fijación interna. Las grandes deformidades dolorosas se tratan con osteotomías correctoras, aunque el porcentaje de recurrencia es elevado. 1,5-7
La displasia fibrosa poliostótica severa con dolores óseos se trata con bifosfonatos. Datos preliminares sugieren que alivian el dolor, reducen la frecuencia de las fracturas patológicas y enlentecen la evolución de la enfermedad ósea. Los bifosfonatos se unen a la superficie de los cristales de hidroxiapatita e inhiben la disolución de dichos cristales. Tienen otros efectos como la reducción de la producción de proteínas y enzimas lisosomales por los osteoclastos y de las unidades de remodelación de hueso neoformado. Los nuevos bifosfonatos (alendronato, pamidronato, reisendronato y tiludronato) son poderosos inhibidores de los osteoclastos con mínimos efectos en la mineralización ósea. 1,7-9
Hasta el presente no existe tratamiento contra el problema molecular específico (inapropiada activación de la subunidad Gsa) de esta enfermedad. Las complicaciones osteomioarticulares dependen del hueso afectado y de la localización específica: fracturas patológicas, osteomielitis secundaria, neuropatías compresivas, contractura de Volkman, distrofia simpaticorrefleja, miositis, osificación ligamentosa y pseudoartrosis. La más temida es la degeneración sarcomatosa posirradiación y puede ocurrir muerte súbita.5, 7 De no ocurrir una transformación maligna, el pronóstico es bueno con grados variables de deformidades.
Summary
Diagnosis and follow-up of a patient with McCune-Albright
The study of the diagnosis and follow-up of McCune-Albright´s disease in a male patient aged 8 years affected by this rare entity was conducted.
Key words: McCune-Albright disease, diagnosis, supplementary tests, radionuclide imaging, bone scintigraphy.
Résumé
Diagnostic et suivi évolutif d'un patient atteint de la maladie de McCune-Albright
Une étude de diagnostic et suivi de la maladie de McCune-Albright, effectué chez un patient masculin âgé de 8 ans et atteint de cette rare entité, est présentée.
Mots clés: maladie de McCune-Albright, diagnostic, tests complémentaires, images par radionucléides, scintigraphie osseuse.
Referencias bibliográficas
1. Frassica FJ. Fibrous dysplasia. Disponible en URL: http://www.ortho.hyperguide.com/tutorial/tumors (Feb 2005).
2. Kairemo KJA, Verho S, Dunkel L. Imaging of McCune-Albright using bone single photon emission computed tomography. Eur J Pediatr. 1999; 158:123-6.
3. Shenker A, Weinstein LS, Moran A, Pescovitz OH, Charest NJ, Boney CM, et al. Severe endocrine and nonendocrine manifestations of the McCune-Albright syndrome associated with activating mutations of stimulatory G protein Gs. J Pediatr. 1993;123:509-18.
4. Santos Silva E, Lumbroso S, Medina M, Gillerot Y, Sultan C, Sokal EM. Demonstration of McCune-Albright mutations in the liver of children with high ?GT progressive cholestasis. J Hepatology. 2000;32:154-8.
5. Boston BA. McCune-Albright syndrome. Disponible en URL: http://www.emedicine.com/ped/topic 1386.htm (Dic 2004).
6. Farber A, Sponseller PD. Fibrous dysplasia. Disponible en URL: http://www.ortho.hyperguides.com/tutorial/tumors/ (Feb 2005).
7. Uwaifo GI, Sarlis NJ. McCune-Albright síndrome. Disponible en URL. http: www.emedicine.com/ped/topic 3194.htm (Jun 2004).
8. Lane JM, Einhorn TA, Kaplan F. Paget´s disease. Disponible en URL: http://www.ortho.hyperguides.com/tutorial/tumor/paget/default.asp (Ene 2005).
9. Frassica FJ. Paget´s disease. Disponible en URL: http://www.ortho.hyperguide.com/tutorial/basic_science/paget´s_disease/default.asp (Sep 2004).
Recibido: 10 de febrero de 2005. Aprobado: 23 de junio de 2005.
Dr. Luis Oscar Marrero Riverón. Complejo Científico Ortopédico Internacional
¨Frank País¨. Ave. 51 No. 19603 entre 196 y 202. La Lisa, Ciudad de La Habana, Cuba. CP11500. E-mail: nuclear@fpais.sld.cu
1 Especialista de II Grado en Ortopedia y Traumatología. Profesor Asistente. Diploma SICOT. Jefe del Departamento de Medicina Nuclear.
2 Especialista de II Grado en Radiología. Jefe del Departamento de Radiología.
3 Especialista de I Grado en Pediatría.
4 Especialista de II Grado en Ortopedia y Traumatología. Profesor Asistente.
5 Especialista de I Grado en Ortopedia y Traumatología.
6 Especialista de I grado en Ortopedia y Traumatología. Profesor Asistente.

