










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Oftalmología
versión On-line ISSN 1561-3070
Rev Cubana Oftalmol v.12 n.1 Ciudad de la Habana ene.-jun. 1999
Centro Provincial de Retinosis Pigmentaria. Camagüey
Herencia de la retinosis pigmentaria en la provincia Camagüey
Elisa Dyce Gordon,1 Yolanda Mapolón Arcendor2 y Beatriz Dyce Gordon3Resumen
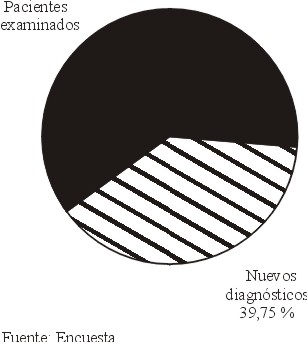
Con el objetivo de clasificar a los pacientes con Retinosis Pigmentaria y a sus respectivas familias según la herencia y exponer el valor de dicha clasificación, se realizó un estudio descriptivo con 354 individuos afectados, distribuidos en 191 familias camagüeyanas. A través de entrevistas y la confección e interpretación del árbol genealógico se obtuvieron los datos necesarios. Se realizó estadística descriptiva con pruebas de chi-cuadrado y de probabilidad estadística. El 36,65 % de las familias estuvieron representadas por los casos con herencia no definida (simple) seguidas por las herencias autosómica recesiva (27,75 %) y autosómica dominante (24,60 %), esta última con el 87 % de penetrancia. Estadísticamente significativa fue la asociación de la consanguinidad con las herencias recesivas ( p < 0,05 ). De 581 pacientes examinados, se realizaron 231 nuevos diagnósticos (39,75 %). El hecho de conocer los modos de herencia de la Retinosis Pigmentaria en cada paciente y familia es de gran valor para el pesquisaje de individuos afectados y para la prevención de la enfermedad.Descriptores DeCS: RETINITIS PIGMENTOSA/ genética; RETINITIS PIGMENTOSA/prevención & control.
La Retinosis Pigmentaria (RP), caracterizada clínicamente por la disminución progresiva del campo visual y su evolución hacia la ceguera,1,2 es la más común de las retinopatías de origen genético,1 representada por tres de los cuatro modos de herencia mendeliana.3,4 La herencia materna (mitocondrial) también ha sido reportada.3,5
Constituye un problema de salud en la población cubana a causa de su prevalencia, estimada en 1 de cada 4 082 habitantes6 y a su carácter incurable, ya que el tratamiento actual está encaminado a mejorar el estado visual del paciente y controlar su evolución; por lo que se hace tan necesaria su prevención y detección precoz.
Con esta investigación se pretende clasificar a los pacientes con RP según los modos de herencia de la enfermedad, relacionándolos con los sexos, razas, edad de aparición y consanguinidad, así como exponer el valor que tiene la identificación de los diferentes patrones hereditarios para perfeccionar su pesquisaje y prevención a través del asesoramiento genético (AG) y el diagnóstico prenatal (en el futuro).
Se realizó un estudio descriptivo sobre la herencia de la RP, en el Centro de Retinosis Pigmentaria de la provincia Cama-güey, desde el 21 de octubre de 1991 hasta el 27 de octubre de 1997. La población estudiada estuvo compuesta por 354 individuos afectados por esta enfermedad, distribuidos en 191 familias camagüeyanas.
A todos los pacientes se les llenó una encuesta en la que se recogieron sus nombres y apellidos, dirección particular, número de historia clínica, sexo, raza y edad de inicio de la enfermedad. Se les confeccionó además el árbol genealógico para establecer los diferentes patrones hereditarios, analizar la consanguinidad y penetrancia del gen.
Para identificar cada patrón hereditario se tuvieron en cuenta los criterios clásicos, más las consideraciones desde el punto de vista práctico tomadas como acuerdos por los genetistas en el marco del IV Taller Nacional de Retinosis Pigmentaria celebrado en diciembre de 1993 en Santiago de Cuba, que son:
RP Autosómica dominante (AD).
- Como mínimo dos generaciones de afectados con transmisión vertical.
- Transmisión de padre a hijo.
- Cada individuo tiene al menos un padre afectado: puede ocurrir un salto generacional.
RP. Autosómica recesiva (AR).
- Dos ó más hermanos afectados, aún con síntomas referidos.
- Dos hermanos varones afectados, hijos de madre normal sin signos de portadora, es decir, con fondo de ojo electrorretinograma normal.
RP Recesiva ligada al cromosoma X (RLX)
Se considerará aquella que reúna los criterios clásicos.
RP con herencia no definida simple (NDS).
Caso aislado, sin signos de consanguinidad parental y sin signos de portadora en madre, hermanas o hijas.
RP con herencia no definida múltiple (NDM).
Familias con al menos dos miembros enfermos que no encuadran dentro de un patrón mendeliano específico.
Caso desconocido
Cuando el individuo con RP sea un hijo adoptivo o desconozca de su familia.
Después de establecer el patrón hereditario en cada caso, se procedió a ofrecer AG e identificación de los individuos con riesgo de padecer la enfermedad. Se les indicó estudio oftalmológico (fondo de ojo, perimetría y electrorretinograma) si ellos así lo deseaban.
La penetrancia del gen en las familias con herencia AD, se calculó en términos matemáticos como:
Individuos afectados
Penetrancia = ___________________ x 100
Individuos Portadores obligados
afectados + del gen simple
La edad de comienzo de la enfermedad (debut) se clasificó según la escuela cubana del profesor Orfilio Peláez Molina en:
Debut precoz: Inicio antes de los diez años.
Debut juvenil: Inicio entre 10 y 20 años.
Debut tardío: Inicio a los 21 años o más.7
Los datos fueron procesados por el paquete de programa estadístico Microstat. Se realizaron pruebas de chi-cuadrado y significación estadística para buscar asociación entre determinadas variables.
Los resultados fueron presentados en tablas de distribución de frecuencias y de contingencia así como en figuras para su análisis y discusión.
Resultados
La mayoría de los pacientes tenían un patrón de herencia bien definido (n = 258; 72,87 %). En las familias predominó el grupo constituido por los casos no definidos simples, seguidos por las herencias AR y AD (tabla 1), esta última con un grado de penetrancia del 87 %.| Modos de Familias | | | ||
| | | | | |
| AD | | | | |
| AR | | | | |
| RLX | | | | |
| Sin patrón hereditario definido | ||||
| NDS | | | | |
| NDM | | | | |
| Desconocida | | | | |
| Total | | | | |
Se encontró un ligero aumento del número de pacientes del sexo masculino (tabla 2) así como un predominio de la raza blanca (tabla 3).
| Sexos | ||
| Modos de herencia | | |
| AD | | |
| AR | | |
| RLX | | |
| NDS | | |
| NDM | | |
| Desconocida | | |
| Total | | |
| Razas | |||
| Modos de herencia Blanca Negra Mestiza | |||
| AD | | | |
| AR | | | |
| RLX | | | |
| NDS | | | |
| NDM | | | |
| Desconocida | | | |
| Total | | | |
Las edades de inicio de la enfermedad fueron variables; el debut precoz fue el más frecuente, seguido por el tardío (tabla 4). En las familias con herencia definida (AD, AR y RLX) se encontró mayor consanguinidad entre aquellas con herencia recesiva (autosómica y ligada al sexo), con una dependencia estadística altamente significativa, con un 99 % de confiabilidad (fig. 1).
| Modos de herencia Debut | |||
| | | | |
| AD | | | |
| AR | | | |
| RLX | | | |
| NDS | | | |
| NDM | | | |
| Desconocida | | | |
| Total | | | |
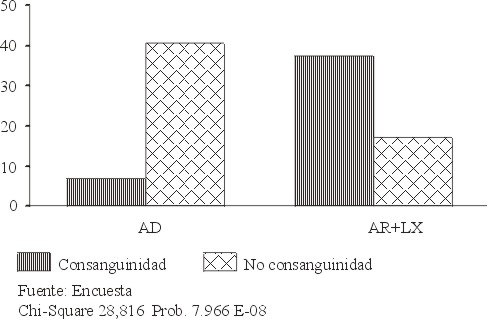
Con el establecimiento del patrón hereditario en cada familia, la identificación de individuos con riesgo de padecer la enfermedad y su estudio oftalmológico, se diagnosticaron 231 nuevos casos de RP (figura 2).
Discusión
Quedó manifiesto el carácter hereditario de la RP en las familias estudiadas. Al igual que en otros reportes de la literatura entre los patrones hereditarios mendelianos, se destaca la herencia AR con alta frecuencia de consanguinidad.1,8 En el caso de la herencia AD la penetrancia del gen resultó ser similar a la reportada por Kaplan y cols, en 19901 y cercano al 90,6 % encontrado por Jay en 1982.4 Posiblemente ascienda más su valor en el futuro, en la misma medida en que se profundice en el estudio de los familiares.El gran número de pacientes con herencia no definida (simple y múltiple) no excluye la base genética en ellos, ya que pudieran representar casos familiares con penetrancia reducida o leve expresión del gen en algunos individuos, así como nuevas mutaciones.
En la literatura se reporta un predominio de esta enfermedad en el sexo masculino a causa de la contribución de la herencia RLX.3 En el presente estudio, el ligero aumento del sexo masculino no está relacionado con este modo de herencia, ya que sólo está presente en tres familias representadas por cinco hombres y una mujer. En los casos no definidos ambos sexos más o menos se afectaron por igual.
El predominio de la enfermedad entre personas blancas es independiente de la composición étnica de la población camagüeyana, pues se han obtenido iguales resultados incluso en las provincias más orientales del país.6
Las diferentes edades de inicio de la enfermedad son una expresión de su gran heterogeneidad clínica y genética1,3 representada por diferentes subtipos clínicos y genéticos que difieren en la edad de comienzo y su evolución.1
Por todo lo anterior concluimos que a través de la identificación de los diferentes modos de herencia de la RP fueron pesquisados de forma organizada un gran número de personas afectadas por la enfermedad, aún cuando se encontraban asintomáticos o con síntomas tan leves que habían pasado inadvertidos.
Es por tanto necesario continuar el estudio de los familiares de los pacientes con herencia no definida, tanto simple como múltiple, para identificar nuevos pacientes. Es necesario también advertir a la población acerca del efecto nocivo para su descendencia del matrimonio entre parientes a causa del riesgo de presentación de enfermedades hereditarias, como es el caso de la RP.
Resumen
A descriptive study of 354 affected individuals distributed in 190 families from Camagüey was conducted aimed at classifying those patients with retinitis pigmentosa and their families according to inheritance and at showing the value of such classification. The necessary data were obtained by interviews and genealogical analysis. A descriptive statistics was presented based on chi square test and statistical probability test. 36,65 % of the families were represent by the cases with indefinite (simple) inheritance followed by recessive autosomal inheritances (27,75 %) and dominant autosomal inheritance (24,60 %). The latter with 87 % of penetrance. The association of consanguinity with the recesive inheritances was statistically significant (p < ,005). 231 new diagnosis (39,75 %) were made among the 581 patients who were examined. Knowing the ways of inheritance of retinis pigmentosa of each patient and this family is very important for screening the affected individuals and for preventing the disease.Subject headings: RETINITIS PIGMENTOSA/ genetics; RETINITIS PIGMENTOSA/prevention & control.
Referencias Bibliográficas
1. Kaplan J, Bonneau D, Frézal J, Munnich A, Dufier JL. Clinical and genetic heterogeneity in retinitis pigmentosa. Hum genet 1990;85:635-42.
2. Peláez O, Palmero A, Pérez RM, Salcedo M. El deterioro visual y su relación con los años de evolución de la Retinosis Pigmentaria. Rev Cubana Oftalmol 1992;4(1):66-71.
3. Haim M. Prevalence of retinitis pigmentosa and allied disorders in Denmark. III. Hereditary pattern. Acta Ophthalmol 1992;70:615-24.
4. Haim M. Retinitis pigmentosa: problems associated with genetic classification. Clin Genet 1993;44:62-70.
5. Dryja TP, Li T. Molecular genetics of retinitis pigmentosa. Hum Mol Genet 1995;4:1739-43.
6. Sarmiento J. Experiencia cubana en la Retinosis Pigmentaria. 1er Simposio Internacional de Retinosis Pigmentaria. Libro de resúmenes. La Habana: Palacio de las Convenciones, 28 nov/1ro dic 1994:12.
7. Peláez O, Herrera M, Palmero A, Pérez RM, Martínez C, Ramos E. Clasificación de la Retinosis Pigmentaria según la escuela cubana del profesor Orfilio Peláez Molina. 1er Simposio Internacional de Retinosis Pigmentaria. Libro de resúmenes. La Habana: Palacio de las Convenciones, 28 nov/1ro dic 1994:25.
8. Kar B, John S, Kumaramanickavel G. Retinitis pigmentosa in India: A genetic and segregation analysis. Clin Genet 1995;47(2):75-9.
Recibido: 17 de marzo de 1998. Aprobado: 5 de abril de 1999.
Dra. Elisa Dyce Gordon. Centro Provincial de Retinosis Pigmentaria. Camagüey.
1 Especialista de II Grado en Genética Clínica. Asistente.
2 Especialista de I Grado en Oftalmología.
3 Especialista de I Grado en Genética Clínica.

