










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Oftalmología
versión On-line ISSN 1561-3070
Rev Cubana Oftalmol v.12 n.2 Ciudad de la Habana jul.-dic. 1999
Instituto de Neurología y Neurocirugía
Alteraciones en el electrorretinograma de los pacientes con distrofia muscular de duchenne y su relación con las deleciones del gen responsable de la enfermedad
Mayra Rodríguez Hernández,1 Rosaralis Santiesteban Freixas,2 Santiago Luis González,3 Rosa Guerra Badía,4 Marta Francisco Plasencia,5 Magdalena Carrero Salgado,6 Yuset Montejo Pujadas7 y Daisy Chi5RESUMEN: La distrofia muscular tipo Duchenne es la más común y grave de las distrofias musculares, que afecta a uno de cada 3,500 varones nacidos vivos. La alta frecuencia con que han sido reportadas alteraciones en el electrorretinograma (ERG) en pacientes con distrofia muscular de Duchenne (DMD), demuestra una posible relación entre la actividad eléctrica de la retina y las mutaciones presentes en el gen DMD que codifica para la proteína distrofina, responsable del fenotipo DMD. La mutación más común del gen DMD son las deleciones del 70 % de los pacientes, y se presentan con una gran heterogeneidad al igual que el ERG, por lo que nos propusimos comparar estas variables para conocer si están o no relacionadas. Se estudiaron 22 pacientes con manifestaciones clínicas de DMD desde el punto de vista molecular y electrorretinográfico para lo cual se utilizó las técnicas de reacción en cadena de la polimerasa para chequear 18 regiones del gen, y el registro electrorretinográfico mesópico. El 69 % de los casos presentaron deleciones, las cuales varían en extensión y ubicación de las cuales el 80 % se encontraba en el extremo 3' del gen. Predominó el ERG de tipo negativo con marcada disminución de la onda B y A en límite inferior normal o también disminuida, con relación b/a menor de uno. Este tipo de ERG, estuvo relacionado con la presencia de deleción (p= 0,027) y su ubicación en el extremo 3' del gen (p= 0,03), con un nivel de significación de 0,05, por lo que se recomienda el estudio electrorretinográfico como un elemento para reforzar el diagnóstico y sospechar el sitio de la deleción.
Descriptores DeCS: DISTROFIA MUSCULAR; ELECTRORRETINOGRAFIA.
La distrofia muscular tipo Duchenne es la más común y grave de las distrofias musculares que afecta a uno de cada 3 500 varones nacidos vivos.1 Existe una forma menos grave de la enfermedad conocida como distrofia muscular de Becker.2 En ambas la causa está dada por alteraciones moleculares en la región Xp 21,1-21,33 y su fenotipo no incluye manifestaciones oftalmológicas evidentes que provoquen deterioro visual. Sin embargo, las notificaciones previas de alteraciones en el electrorretinograma (ERG) de pacientes con distrofia muscular de Duchenne (DMD),4-6 demuestra una posible relación entre la actividad eléctrica de la retina, que este biopotencial recoge, y las mutaciones presentes en el gen que codifica para la proteína distrofina, gen DMD, que es responsable del fenotipo DMD.7-10
Se han descrito 8 isoformas de la proteína distrofina,11 cuyas diferencias radican en el tamaño y secuencia con respecto a la forma básica, producto de procesos moleculares como:
- El usos de diferentes promotores para la proteína en diferentes tejidos.
- El "splicing" diferencial del RNA mensajero.
- Diferentes sitios de poliadenilación.
En 1994, se reportaron los primeros estudios realizados en el Instituto de Neurología y Neurocirugía (INN) con 20 pacientes DMD, midiendo los cambios morfológicos del ERG, y se encontró que la mayoría presentaban un ERG de tipo negativo, o subnormal con gran disminución de la amplitud de la onda A sin relación entre los resultados electrorretinográficos, la clínica y el estado de la enfermedad.5 Esto hizo plantear, desde entonces, una posible afectación en la transmisión sináptica en el nivel de la capa externa plexiforme de retina, cuya alteración pudiera justificar las anormalidades en el ERG de estos pacientes, lo que estuvo avalado por los trabajos de Miyatake12 quien había reportado la presencia de distrofina en dicha capa en ratones con la enfermedad.
En 1996, se presentaron los resultados obtenidos en una pequeña población estudiada con métodos de biología molecular en el INN, sugiriendo una posible relación entre la región en que se encuentra la deleción y el tipo de ERG.8
Hoy se conoce que en la retina se presentan 3 isoformas de la distrofina (Dp), caracterizadas por presentar diferentes pesos moleculares y secuencias únicas en el extremo amino terminal, estas isoformas se nombran Dp427, Dp260 y Dp71. Desde el punto de vista funcional las isoformas Dp260 y Dp71 presentan mayor importancia y entre ellas se diferencian en la secuencia del carbono terminal y en la región en que se encuentran: la Dp260 se encuentra en la capa plexiforme externa de la retina y la Dp71 en la membrana limitante interna y en los vasos sanguíneos.9
La mutación más común del gen DMD son las deleciones del 70 % de los pacientes,13 que se presentan con una gran variabilidad en cuanto a localización y extensión en el gen, tanto en pacientes con DMD como con Distrofia Muscular de Becker (DMB).
La heterogeneidad del comportamiento del ERG y las deleciones en estos pacientes motivó su estudio, para definir si existe alguna relación entre ellos.
En este trabajo nos propusimos, aumentar el número de pacientes y las posibilidades de detección de deleciones al 98 %, para corroborar la posible relación entre el ERG y la presencia o no de deleción, y la zona en que ella se encuentra. Estos resultados podrían contribuir a la comprensión del comportamiento de la distrofina en la retina y además conocer la posibilidad de utilizar este estudio como un complemento diagnóstico en tan controvertida enfermedad.
Método
Se estudiaron 22 pacientes con características clínica o fenotípicas de DMD avalada por historia de fuerza muscular disminuida, seudohipertrofia gemelar, enzimas creatinfosfoquinasa (CPK) elevadas, biopsia y electromiograma miopático.Se realizó estudio neuroftalmológico y el registro electrorretinográfico se llevó a cabo en un equipo Handaya con estímulos de 5 y 20 joules y 8 promediaciones con intervalo inter estímulos de 3 s, constantes de tiempo de 300 ms. No se usó medicación midriática. Se evaluaron los registros según la norma establecida para cada edad en población sana del propio laboratorio, según latencia y amplitud.
Se utilizó la clasificación de Karpe y Henkes para evaluar el registro del ERG (normal, supernormal, subnormal, negativo e inexistente). Además se midió la relación b/a del ERG (normal 1,5 o más).
El patrón de herencia de la enfermedad en cada familia se definió de acuerdo con el árbol genealógico.
El estudio molecular se realizó mediante el método múltiple de reacción en cadena de la polimerasa (PCR) descrito por Chamberlain y otros 198914 y Beggs y otros 1990,15 con lo cual se chequean 18 regiones de gen DMD que coinciden con los exones más susceptibles de sufrir deleciones, desde el promotor del gen hasta el exón 60 (el gen DMD tiene 79 exones).
Los resultados se observan por ausencia o presencia de banda en el patrón electroforético en un gel de agarosa al 2 %.
Se comparó el ERG con la presencia de deleción y con la región en que se ubicó la deleción en el gen. Se usó para ello el método estadístico de chi cuadrado, con un nivel de significación de 0,05.
Resultados
Se estudiaron 22 varones comprendidos entre las edades de 4 y 16 años. En 15 de estos pacientes se detectó una deleción como causa de la enfermedad, para el 68 %, por lo que de acuerdo con la eficiencia del método, que es del 98 % de deleción, se pudo decir que el 69 % de los casos presentaron una deleción. Estas deleciones variaron en su extensión desde un exón hasta 4 exones y en cuanto a ubicación, el 80 % se situó en el extremo 3 del gen involucrando fundamentalmente los exones desde el 44 hasta el 51. De los pacientes que presentaron deleción ésta se extendió más allá del exón 60.En la muestra en general predominó el ERG de tipo negativo con una relación b/a < 1, lo que estuvo relacionado (p=0,027) con la presencia de deleciones. El ERG fue anormal en 14 pacientes (64 %), con marcada disminución de la onda B y A en límite inferior normal o también disminuida, siempre con relación b/a menor de 1, en los 14 casos. Estos tipos de ERG se vieron sólo en los casos cuya deleción estaba en el extremo 3', con dependencia entre el ERG alterado y la presencia de deleción (p < 0,003) (Fig.). Sólo un caso con deleción en el extremo 3' del gen tuvo ERG normal y se correspondió con una distrofia muscular de Becker (DMB), cuya evolución clínica es muy diferente al resto de los pacientes, por lo que no es raro que el comportamiento de ERG también difiera de lo esperado.
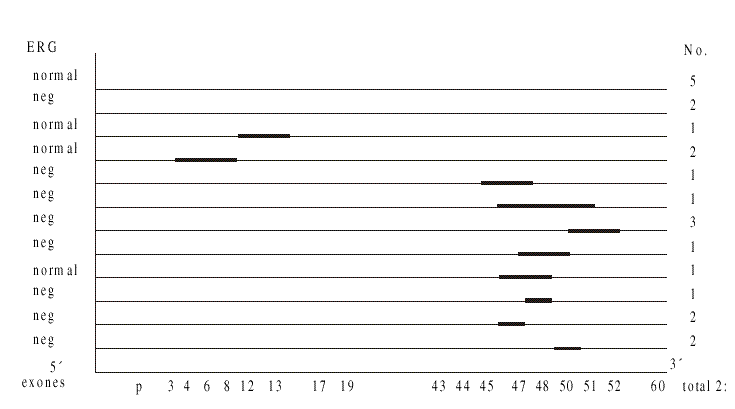
FIG. Representación esquemática del gen DMD 5' del gen hasta extremo 3'.
Las deleciones se representan como líneas horizontales sobre los exones promotores del gen. En el borde izquierdo se relacionan los resultados del ERG, normal o negativo. En el extremo derecho se indica el número de pacientes para genotipo y fenotipo relacionado.
En el extremo 5' del gen sólo 3 pacientes presentaron deleción, nunca se incluyó el promotor del gen y en ellos el ERG fue normal.
Discusión
La DMD se presenta, generalmente, en niños varones a causa del tipo de herencia: ligada al sexo. Tiene un carácter irreversible y los primeros síntomas se presentan alrededor de los 4 años de edad.De los 22 pacientes estudiados, 15 tuvieron deleción. La presencia de la deleción en un elevado porcentaje de esta muestra, como causa de la enfermedad, coincide con lo expresado por otros autores13 y con lo notificado previamente por nosotros en la población cubana.16 Esto permite conocer con qué frecuencia se puede detectar de forma directa la mutación en la población afectada, lo que puede aplicarse para los diagnósticos presintomáticos, de portadoras y casos prenatales de la enfermedad.
En 15 de los pacientes que tuvieron deleción, 11 presentaban alteración del ERG con disminución preferencial de la onda B, y 4 con ERG normal, por lo que al comparar estas variables podemos plantear que hay una dependencia entre el ERG alterado y la presencia de deleción (p=0,027). Piller et al. 19937 reporta resultados similares con respecto al ERG, sin embargo no encuentra relación entre las variables por nosotros comparadas. Esto pudo estar determinado por la muestra que utiliza de sólo 8 pacientes.
En la DMD el extremo 3' del gen es el más susceptible de sufrir mutaciones, y los exones ubicados entre el 45 y 51 son los más propensos a estar ausentes, por lo que se conoce este sitio como "hot spot" por su alta frecuencia de mutación.17 Nuestro estudio reafirma que la ubicación de las deleciones es más común en el extremo 3', pues se encontró que en el 80 % de los casos la deleción ocurría en esa región. El comportamiento heterogéneo en cuanto a longitud de la deleción fue también el esperado.18
Los exones más distales chequeados fueron el promotor del gen en el extremo 5'y el exón 60 en el extremo 3'; ambos estuvieron presentes en todos los pacientes, por lo que la deleción nunca se extendió hasta los extremos del gen. Sin embargo, no podemos descartar la posibilidad de que en algunos de los pacientes en los que no se detectó deleción, estuviera ausente algún sitio posterior al exón 60 que pudiera o no extenderse hasta la región más distal del extremo 3'del gen.
La dependencia entre la ubicación de las deleciones en el extremo 3'del gen y las alteraciones en el ERG puede ser explicado ya que las isoformas de la distrofina con una contribución funcional importante en la retina son, las isoformas Dp260, cuya transcripsión comienza por un promotor localizado en el intrón 29 del gen DMD, y la Dp71 que presenta el promotor en el exón 63,9 por tanto los exones del extremo 5' del gen no son transcritos para codificar estas isoformas de la distrofina: por esto, una deleción en el extremo 5'del gen, puede no contribuir a la alteración del ERG en estos pacientes.
A pesar de haber encontrado relación entre presencia de deleción y alteración del ERG, 2 pacientes que no presentaban deleción tuvieron ERG negativo. Esto lo podemos atribuir a que la deleción se encuentre en una región no detectada por el método utilizado (2 % de las deleciones no son detectadas), pero que altere la transcripción de una de las isoformas expresadas en la retina, como la Dp71, que su expresión depende de los exones posteriores al 63 y el método utilizado sólo detecta hasta el exón 60.
La presencia de la Dp260 en la capa plexiforme externa de la retina y de la Dp71 en la membrana limitante interna y en los vasos sanguíneos, hace pensar en una contribución esencial pero diferente desde el punto de vista funcional de cada una de ellas, por lo que podría esperarse alguna diferencia en cuanto a la característica del ERG entre los pacientes que presenten una alteración en las isoformas Dp260 y la Dp71. Por tanto, al profundizar en el estudio electrorretinográfico, se podría encontrar diferencias entre los pacientes que presentan ERG alterado y una deleción anterior al exón 63 (11 pacientes) y los pacientes que tienen ERG alterado, y por no detectarse mutación, pudieran tener una deleción posterior al exón 63 (2 pacientes). La comprobación de esta hipótesis contribuiría al conocimiento en cuanto a la función de cada una de estas isformas de la distrofina en la retina, además permitiría una relación entre el ERG y la región del gen que está afectada, útil a la hora de orientar los estudios moleculares.
Con el aumento de la eficiencia del método utilizado para detectar mutaciones en el gen DMD y del número de la muestra, pudimos corroborar la relación que existe entre las deleciones presentes en dicho gen y el ERG de los pacientes con DMD. Además sobre la base de nuestros resultados recomendamos profundizar en el estudio electrorretinográfico, como un elemento para reforzar el diagnóstico y sospechar el sitio de la deleción en el gen.
En otro sentido se debe prestar especial atención a los casos que no tuvieron deleción y presentan ERG normal, pues pudieran no corresponder a una DMD.
SUMMARY: Duchenne muscular dystrophy is the most common and serious of all muscular distropies affecting one in 3 500 males born live. The high frequency of reported alterations in electroretinographies of patients with Duchenne muscular dystrophy (DMD) proves that there is a possible relationship between the electrical activity of retina and the DMD gen mutations, a gen which codes for dystrophin protein responsible for DMD phenotype. The most common DMD gen mutation is deletions in 70 % of patients and they occured with a high heterogeneity as it happens in electro retinography; so we intended to compare these variables to find out whether thy are related. 22 patients with DMD clinical manifestations were studied from the molecular and electroretinographic viewpoints. Polymerase chain reaction technique was used to check 18 gen regions and the mesopic electroretinographic recording. 69 % of cases had deletions which varied in extension and location; 80 % of deletions was found in gen 3' extreme. ERG of negative type with marked reduction of B and A waves in normal lower limit or also decreased were predominant; b/a relation was under 1. This type of ERP was related to deletion (p=0,027) and its location in gen 3' extreme (p=0,03). The level of significance was 0,05, so we recommend the electroretinographic study as a way of strenghtening diagnosis and detecting the suspected deletion locus.
Subject headings: MUSCULAR DYSTROPHY; ELECTRORETINOGRAPHY.
Referencias Bibliográficas
1. Duchenne GA. Recherches sur la paralysie musculaire pseudohypertrophique ou paralysie myo-sclerosique. Arch Gen Med 1868;5(25):179-209.
2 .Hidalgo PC. Bases moleculares de las distrofias musculares ligadas al cromosoma X: Duchenne y Bercker. Inicio de una nueva época en la medicina, Medicentro 1989;4(2):254-7.
3. Davies KE, Parson PL, Harper PS, et al. Linkage analysis of two cloned DNA sequences flanking the DMD locus of the short arm of the human X-chromosome. Nucleic Acids Res 1983; 11:2303-12.
4. Fitzgerald KM, Cibis GW, Giambrone SA, Harris DJ. Retinal signal transmission in Duchenne muscular dystrophy: evidence for dysfunction in the photoreceptor/depolarizing bipolar cell pathway. J Clin Invest 1994;93(6):2425-30.
5. Santieste R et al. ERG in Duchenne disease. Proceding simpos. NANOS, 1994.
6. Santiesteban y otros. La retina en la enfermedad de Duchenne. Estudios electrorretinográficos. Rev Cubana Oftalmol 1996;9(2):86-91.
7. Pillers DM, Bulman DE, Weleber RG, Sigesmund DA, Musarella MA, Powell BR, et al. Dystrophin expression in the human retina is required for normal function as defined by electroretino-graphy. Nature Gen 1993;4:82-6.
8. Rodríguez M, Santiesteban R, Ferreira R, Luis S. Correlación entre las deleciones en el gen DMD y las alteraciones en el electrorretinograma de pacientes con distrofia muscular de Duchenne. Rev Cubana Oftalmol 1996;2:92-5.
9. Haward PL, Dally GY, Wong MH, et al. Localization of dystrophin isoform Dp71 to the inner limiting membrane of the retina suggests a unique functional contribution of Dp71 in the retina. Hum Mol Gen 1988; 7(9):1385-91.
10. Pascual SI, Molano J, Pascual-Castroviejo I. Electroretinogram in Duchenne/Becker muscular dystrophy. Pediatr Neurol 1998;18(4):315-20.
11. Ino-ue M, Honda S, Nishio H, Matsuo M, Nakammura H, Yamamoto M. Genotype and electroretinal heterogeneity in Duchenne muscular dystrophy. Exp Eye Res 1997;65(6):861-4.
12. Miyatake M, Milke T, Zhao JE, Yoshiaka K, Uchino M, Usuku G. Dystrophin: localization and presumed function. Muscle Nerve 1991;14:113-9.
13. Lindlof M, Kiuru A, Kaariainen H, Kalimo H, Lang H, Pihko H, et al. Gene deletions in X-linked muscular dystrophy. Am J Hum Gen 1989;44:496-503.
14. Chamberlain JS, Gibbs RA, Rainier JE, Nguyen PN, Caskey CT. Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res 1988;23:11141-56.
15. Beegs AH, Koenig M, Boyce FM, Kunkel LM. Detection of 98-percent of DMD/DMB gene deletion by PCR: Hum Gen 1990;86:45.
16. Rodríguez M, y col. Diagnóstico prenatal y de portadoras de la distrofia muscular de Duchenne. Rev Cubana Pediatr 1996;(1):11-20.
17. Den-Dunnen JT, Gootscholten PM, Bakker E, et al. Topography of the DMD-gene: FIGE-and cDNA analysis of 194 cases reveals 115 deletions and 113 duplications. Am J Hum Gen 1989;45: 835-47.
18. Rodríguez M, Ferreira R, Gayol LA, Luis RS. Caracterización de deleciones en la DMD: su frecuencia en la población cubana. Rev Cubana Med 1996;68(1).Recibido: 1ro. de diciembre de 1998. Aprobado: 12 de julio de 1999.
Dra. Mayra Rodríguez Hernández. Instituto de Neurología y Neurocirugía, Ciudad de La Habana, Cuba.
1 Investigadora Agregada.
2 Especialista de II Grado en Oftalmología. Profesor Auxiliar. Investigadora Titular.
3 Especialista de II Grado en Neurología. Investigador Titular. Director INN.
4 Técnica de Laboratorio.
5 Especialista de I Grado en Oftalmología.
6 Licenciada en Oftalmología y Optometría.
7 Estudiante de 5to. año de Bioquímica de la Facultad de Biología.

