










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Oftalmología
versión On-line ISSN 1561-3070
Rev Cubana Oftalmol v.22 n.2 Ciudad de la Habana jul.-dic. 2009
REVISIÓN BIBLIOGRÁFICA
Manifestaciones oculares en la drepanocitosis
Ocular manifestations in drepanocytosis
Icilany Villares ÁlvarezI; Bárbara Teresa Ríos AraújoII; Julio Dámaso Fernández ÁguilaIII; Marylín Aroche QuintanaIV; Yusimí Fojaco ColinaV
IEspecialista de I Grado en Oftalmología. Especialista de I Grado en Medicina General Integral. Servicio de Retina. Hospital Universitario "Dr. Gustavo Aldereguía Lima". Cienfuegos, Cuba.
IIEspecialista de II Grado en Oftalmología. Servicio de Retina. Hospital Universitario "Dr. Gustavo Aldereguía Lima". Cienfuegos, Cuba.
IIIEspecialista de II Grado en Hematología. Investigador Auxiliar. Asistente. Servicio de Oncohematología. Hospital Universitario "Dr. Gustavo Aldereguía Lima". Cienfuegos, Cuba.
IVEspecialista de I Grado en Oftalmología. Especialista de I Grado en Medicina General Integral. Servicio de Retina. Hospital Universitario "Dr. Gustavo Aldereguía Lima". Cienfuegos, Cuba.
VEspecialista de I Grado en Oftalmología. Especialista de I Grado en Medicina General Integral. Servicio de Retina. Hospital Universitario "Dr. Gustavo Aldereguía Lima". Cienfuegos, Cuba.
RESUMEN
La drepanocitosis es la anemia hemolítica congénita más frecuente en Cuba. Sus manifestaciones clínicas fundamentales aparecen por eventos de oclusión vascular y acortamiento de la vida de los hematíes. En esta enfermedad pueden afectarse todas las estructuras anatómicas del ojo, órgano que ofrece la oportunidad única de observación del proceso vasooclusivo. El signo conjuntival más característico es la aparición de segmentos venulares poscapilares en forma de coma, que se consideran diagnósticos de la enfermedad, aunque no son patognomónicos, porque pueden encontrarse en otras hemopatías. La afectación del segmento anterior se caracteriza por inyección conjuntival, edema de la córnea y precipitados queráticos. Tardíamente hay atrofia y despigmentación del iris. Se ha reportado rubeosis iridiana. En el segmento posterior se ha descrito una gran variedad de lesiones que se producen por vasooclusión y que, en ocasiones, no conducen a la disminución de la visión ni requieren tratamiento específico. La retinopatía proliferativa de la drepanocitosis es más frecuente en enfermos con hemoglobinopatía SC; puede tener regresión espontánea y se diferencia de otras entidades con isquemia retiniana, en que sus lesiones generalmente se localizan hacia la periferia y no afectan la visión central del enfermo.
Palabras clave: Drepanocitosis, manifestaciones clínicas.
ABSTRACT
Drepanocytosis is the most common congenital hemolytic anemia in Cuba. The onset of fundamental clinical manifestations is due to vascular occlusion and shortened red cell life. This disease may affect all the anatomical structures of the eye, the organ that may provide the only opportunity for seeing the vasocclusive process. The most characteristic signal is the onset of coma-like postcapillary venular segments that are considered as diagnosis of the disease, although they are not pathognomonic because they may be found in other hemopathies. The effect of the anterior segment is characterized by conjunctival injection, corneal edema and keratic precipitates. Later on, there is iris atrophy and depigmentation. Iris rubeosis has been reported. There have been described a wide variety of lesions caused by vasocclusion in the posterior segment; occasionally, they neither lead to decreased vision nor require specific treatment. Proliferative retinopathy of drepanocytosis is more frequently found in patients with hemoglobinopathy SC; it may have spontaneous regression and is different from other entities with retinal ischemia in that the lesions are generally located toward the periphery and do not affect the central vision of the patient.
Key words: Drepanocytosis, clinical manifestations.
INTRODUCCIÓN
La drepanocitosis es un grupo de anemias hemolíticas congénitas que incluye a la anemia drepanocítica (AD), la hemoglobinopatía SC (HSC) y la Sß talasemia (Sß tal).
En 1910, James Herrick realizó la primera descripción de la AD, al diagnosticarla en un estudiante de Granada que padecía dolor recurrente, anemia y en cuya sangre observaron hematíes falciformes.1 Posteriormente, en 1957, Ingram demostró que la sustitución de una molécula de ácido glutámico por valina en la sexta posición de la cadena ß de la hemoglobina (Hb), es la que lleva a la formación de una Hb anormal (Hb S, a2 ß2 s val).2,3
El origen de la mutación no está bien documentado. La hipótesis más probable es que en zonas donde la malaria es endémica (norte de África, Arabia, India), los individuos heterocigóticos tuvieron alguna ventaja selectiva con respecto a los homocigóticos, tanto sanos como enfermos, y de esta forma perpetuaron el gen mutado. Posteriormente su distribución al resto del mundo se vio influenciada por el comercio de esclavos.4,5 En la población afro-norteamericana se registra una prevalencia de portadores de 8 a 10 %, y en el oeste africano alcanza el 30 % en algunos países.6
El número aproximado de portadores en Cuba es de 300 000, con una frecuencia 13,2 en negros, 4,1 en mestizos, 0,6 en blancos y 3,08 % en la población en general. Se calcula que existan alrededor de 4 000 enfermos, distribuidos por todo el país, con mayor prevalencia en Ciudad de La Habana y en las provincias del sur de la región oriental.7,8
La polimerización de la Hb S en estado desoxigenado es la base fisiopatológica de la drepanocitosis. Los hematíes se distorsionan, disminuyen su flexibilidad y pueden producir la oclusión de pequeños vasos sanguíneos, así como la oxigenación deficiente de los tejidos. Esto trae como consecuencia las crisis vasooclusivas (CVO) y el acortamiento de la sobrevida de los glóbulos rojos.9 La repetición de las CVO y la oclusión vascular crónica, constante y subclínica, conduce a alteraciones permanentes e irreversibles de los órganos.4
MANIFESTACIONES OCULARES
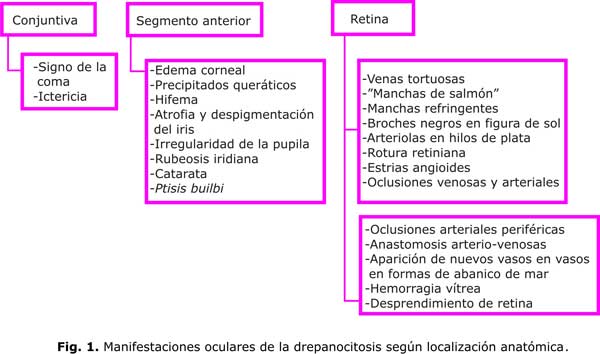
La drepanocitosis puede afectar todas las estructuras anatómicas del ojo (fig.1), órgano que ofrece la oportunidad única de observación del proceso vasooclusivo.
En el año 1930, Cook y otros realizaron la primera descripción de alteraciones en el fondo de ojo asociadas con la drepanocitosis, en un enfermo que padecía hemorragias retinianas, quien falleció por una hemorragia subaracnoidea.10 Edington y Sarkies reportaron, en 1952, dos enfermos que presentaban aneurismas retinianos y hemorragia vítrea.11 En 1966, Welch y Goldberg publicaron una serie de casos, integrados por 55 pacientes homocigóticos para el gen ßs (SS), 34 individuos portadores (AS), 22 doble heterocigóticos (SC) y 38 personas con hemoglobina normal, en los cuales analizaron la frecuencia de las manifestaciones oculares de la enfermedad.12
Conjuntiva
Las alteraciones de la red vascular superficial de la conjuntiva bulbar son comunes. El signo conjuntival más característico de la anemia por células falciformes es la aparición de segmentos venulares poscapilares en forma de coma o sacacorchos, de color rojo oscuro. Se localizan generalmente en la conjuntiva bulbar inferior, cerca del limbo esclerocorneal. Estos segmentos venulares se consideran evidencia en el diagnóstico de la enfermedad de células falciformes, aunque no son patognomónicos porque pueden encontrarse en otras hemopatías como la leucemia mieloide crónica.13
El signo de la coma es más frecuente en la AD que en la HSC y en la Sß tal (AD>HSC>Sß tal).14-16 Esta anomalía no se modifica por la inhalación de oxígeno; es poco frecuente en pacientes con niveles altos de Hb F y desaparece transitoriamente después de las transfusiones. El número de lesiones se ha relacionado con el curso clínico de los enfermos.15 Lima y otros observaron que es más frecuente en pacientes con AD y Hb < 90 g/L y que no tienen influencia la edad, el sexo, el valor de la Hb F, los haplotipos de genes del bloque ß y la a talasemia.16 Se reporta ictericia conjuntival en la mitad de los pacientes.17
Segmento anterior
La afectación del segmento anterior se caracteriza por inyección conjuntival, edema de la córnea, precipitados queráticos, disminución de la presión intraocular con pupilas dilatadas y arrefléxicas. Tardíamente se observa atrofia y despigmentación del iris, irregularidad de la pupila, catarata y, en algunos casos, ptisis bulbo.15 También se ha reportado rubeosis iridiana.18
Las oclusiones vasculares del tracto uveal se han comunicado en la drepanocitosis, lo cual provoca la aparición de atrofias y rubeosis del iris con glaucoma neovascular y oclusiones de los vasos coroideos. Esta alteración está estrechamente relacionada con la retinopatía de la drepanocitosis al igual que las opacidades del vítreo. Acheson reporta atrofia del iris en el 14,7 % de los integrantes de su serie con HSC,19 y Clarkson en el 3,4 % de los que tienen el referido genotipo.20 El hifema en pacientes con drepanocitosis aparece generalmente después de un traumatismo ocular y puede conducir a un glaucoma secundario por trastornos en el drenaje del humor acuoso.21
Segmento posterior
En el segmento posterior se ha descrito una gran variedad de lesiones que se producen por vasooclusión y que, en ocasiones, no conducen a disminución de la visión ni requieren tratamiento específico, porque generalmente se localizan hacia la periferia y no afectan la visión central del enfermo,22 hecho que diferencia la retinopatía falciforme de otras entidades con isquemia retiniana.
La frecuencia de retinopatía en enfermos con drepanocitosis varía en diferentes fuentes consultadas. En una cohorte de 90 pacientes (88 con AD y 2 con HSC), se reporta retinopatía no proliferativa en 21 %, proliferativa en 5,6 y 2,2 % con ambas.23 En un estudio conducido en Mali se detectó en el 68 % de los casos, en el 90 % de los doble heterocigóticos (SC), en el 10 % de portadores (AS) y no fue detectada en ningún SS.24 Otro estudio realizado en África informa 44,2 % de retinopatía, con 26,3 de SC afectados y 11 % de SS.25
Un artículo publicado en el año 2005 expone los resultados del seguimiento realizado durante 20 años a 307 niños con Hb SS y 166 con Hb SC nacidos en Jamaica, en el cual la retinopatía fue tres veces más frecuente en la HSC que en la AD, con una tasa anual de incidencia de 0,5 casos por 100 en los SS y de 2,5 por 100 en los SC.26 En general hay consenso en que, a pesar de ser más graves las complicaciones sistémicas en la AD que las de la HSC, la pérdida visual por retinopatía proliferativa es más común en esta última. Esta anomalía no tiene una explicación clara.27
Retinopatía falciforme no proliferativa
El proceso patológico primario en la retinopatía de células falciformes es la oclusión vascular de la retina periférica. La aparición de arteriolas en hilos de plata puede ser el primer signo. Después de la oclusión surge la hemorragia en "placa de salmón", generalmente periférica, superficial, de forma ovalada y bien delimitada, con destacada coloración rojo anaranjada.28 Estas placas, al reabsorberse, dejan "manchas refrigentes" por depósito de hemosiderina, sin afectación de la agudeza visual. Otra posible evolución de las placas de salmón cuando la hemorragia afecta las capas profundas de la retina es hacia la cicatriz con atrofia coriorretiniana periférica, que adquiere aspecto de "broche negro en figura de sol".
Pueden aparecer, además, venas tortuosas, rotura retiniana en la zona ecuatorial o preecuatorial y estrías angioides.29 Estas últimas son estrías pigmentadas del fondo, semejante a vasos sanguíneos, que afectan las porciones colágenas y elásticas de la membrana de Bruch. Aparecen como consecuencia del depósito de hierro, calcio o ambas14 y en el 50 % de los casos se asocian a trastornos sistémicos como la AD, el rasgo drepanocítico (Hb AS), HSC ß tal, Hb H, a talasemia homocigótica y heterocigótica.29
Las estrías angioides son más comunes en la AD y en enfermos con edad avanzada. Se reportan en alrededor del 2 % de los enfermos con drepanocitosis, pero en individuos homocigóticos (SS) y mayores de 40 años se han demostrado en el 22 % de los casos estudiados.20 Generalmente son asintomáticas, aunque se han descrito casos en que progresan a afectación macular, degeneración disciforme y neovascularización subretinal con afectación de la visión. Se han reportado problemas tales como oclusión en la arteria central de la retina, en la vena retiniana y también en vasos coroideos.29-31
Retinopatía proliferativa de la drepanocitosis
El evento inicial en la patogénesis de la retinopatía proliferativa de la drepanocitosis (RPD) es la oclusión de arteriolas de la retina periférica. El tejido retiniano hipóxico por oclusiones repetidas estimula la angiogénesis mediante la producción de factores de crecimiento vascular, como el factor de crecimiento del endotelio vascular (VEGF) y el factor del crecimiento básico de los fibroblastos (BFGF).32,33
En 1971, Goldberg definió cinco estadios de RPD (fig. 2):34
- Estadio 1: se caracteriza por oclusiones arteriales periféricas.
- Estadio 2: remodelación vascular, formación de anastomosis arteriovenosas periféricas que aparecen como canales vasculares en los límites entre la retina periférica y la no periférica tras la oclusión vascular.
- Estadio 3: aparición de nuevos vasos que muestran una configuración en forma de abanico de mar.
- Estadio 4: se caracteriza por la presencia de hemorragia vítrea de grado variable.
- Estadio 5: hay tracción del vítreo y desprendimiento de la retina.
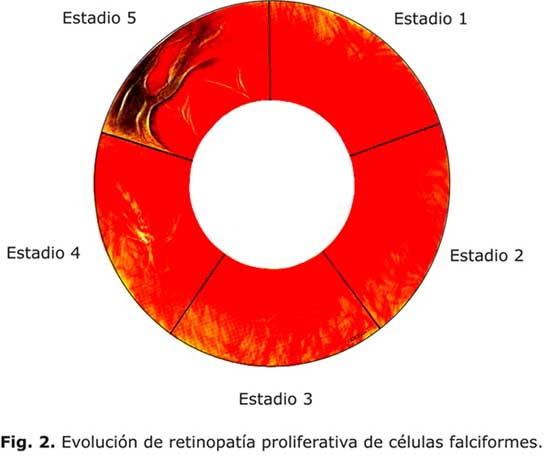
La RPD es más frecuente en enfermos con HSC y con Sß tal que en aquellos con AD. La prevalencia es mayor en la segunda mitad de la tercera década de la vida, en el sexo masculino y en individuos con bajos niveles de Hb F.15,26,35,36
La evaluación periódica por un oftalmólogo de los enfermos con drepanocitosis y la detección de la retinopatía en sus estadios iniciales es esencial para conservar la visión de estos pacientes. Como se reportan casos de regresión espontánea de la RPD, la fotocoagulación con láser se reserva a algunos casos con mal pronóstico.37
REFERENCIAS BIBLIOGRÁFICAS
1. Herrick JB. Peculiar elongated and sickle shaped red blood corpuscles in a case of severe anemia. Arch Inter Med. 1910;6:517.
2. Embury SH Anemia de células falciformes y hemoglobinopatías relacionadas. En: Bennett JC, Plum F, editores. Tratado de Medicina Interna. Ciudad de La Habana: Editorial Ciencias Médicas. 1996. p. 1013-26.
3. Benz EJ. Genotypes and phenotypes. Another lesson from the hemoglobinopathies. N Engl J Med. 2004;351(15):1490-2.
4. Colombo B, Svarch EG, Martínez G. Genética y clínica de las hemoglobinopatías humanas. Ciudad de La Habana: Pueblo y Educación. 1993. p. 146-95.
5. Pagnier J, Mears JD, Dunda Belkhodia O, Shafer Rego KE, Beljord C, Nagel RL, et al. Evidence for the multicentre of the sickle cell hemoglobin gene in Africa. Proc Natl Acad Sci USA. 1994;81:1771-3.
6. Sear DA. Sickle Cell Trait. En: Embury Slt, Hebbel RP, Mohandes N, Steinberg NH, editors. Sickle cell disease. Basic principles and clinical practice. New York: Raven Press. 1994. p. 381-3.
7. Vidal H, Hernández A, Colombo B. Genetic and clinical relevance of hemoglobin screening: results from a survey in a pediatric hospital. Clin Genet. 1974;5:31-5.
8. Svarch E, Hernández P, Ballester JM. La drepanocitosis en Cuba. Rev Cubana Hematol Inmunol Hemoter. 2004.20(2):[aprox.6.p]. [Consultado: 11 de diciembre de 2007]. Disponible en:http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892004000200009&lng=es&nrm=iso.
9. Rudgers G. Overview of pathophisiology and rationale for treatment of sickle cell anemia. Sem Hematol. 1997;34:2-7.
10. Cook WC. A case of sickle-cell anemia with associated subarachnoid hemorrhage. J Med. 1930;11:541.
11. Edington GM, Sarkies JWR. Two cases of sickle cell anaemia associated with retinal microaneurysms. Trans R Soc Trop Med Hig. 1952;46:59-62.
12. Welch RB, Goldberg MF. Sickle-cell hemoglobin and its relation to fundus abnormality. Arch Ophthalmol. 1966;75:353-62.
13. Marín J, Saavedra S, Sanz M, Díaz-Llopis M. Enfermedades de células falciformes y otras hemoglobinopatías. En: Sánchez M, Díaz-Llopis M, Benítez del Castillo JM, Rodríguez MT. Manifestaciones oftalmológicas de las enfermedades generales. Madrid: Elsevier. 2001. p. 202-5.
14. Charache S. Eye disease in sickling disorders. Hematol Oncol Clin N Am. 1996;10:1357-62.
15. Serjeant GR. Serjeant BC. Sickle cell disease. Oxford: Oxford University Press. 2001. p. 315-39.
16. Lima CS, Rocha EM, Silva NM, Sonatti MF, Costa FF, Saad ST. Risk factors for conjunctival and retinal vessel alterations in sickle cell disease. Acta Opthalmol Scand. 2006;84(2):234-41.
17. Akinsola FB, Kehinde MO. Ocular findings in sickle cell disease patients in Lagos. Niger Postgrad Med J. 2004;11:203-6.
18. Spires R. Ocular manifestations of sickle cell disease. J Ophthalmic Nurs Techonol. 1995;14(2):74-7.
19. Acheson RW, Ford SM, Maude GH, Lyness RW, Serjeant GR. Iris atrophy in sickle cell disease. Br J Ophthalmol. 1986;70:516-21.
20. Clarkson JG. The ocular manifestations of sickle cell disease: a prevalence and natural history study. Trans Am Ophthalmol Soc. 1992;90:481-504.
21. Goldberg MF. Sickled erythrocytes, hyphema, and secondary glaucoma. Opthalmic Surg. 1979;10:117-23.
22. Weingeist TA, Liesegang TJ, Grand MG. Retina and vitreous. San Francisco: America Academy of Ophthalmology. 2000. p. 86-90.
23. Babalola OE, Wambebe CO. Ocular morbidity from sickle cell disease in a Nigerian cohort. Niger Postgrad Med J. 2005;12(4):241-4.
24. Traore J, Boitre JP, Bogoreh IA, Traore L, Diallo A. Sickle cell and retinal damage: a study of 38 cases at the African Tropical Ophthalmology Institute (IOTA) in Bamako. Med Trop (Mars). 2006;66(3):252-4.
25. Balo KP, Fany A, Mihluedo H, Djagnikpo PA, Koffi-Gue KB. Retinal involvement in drepanocytosis in Togo. Correlation between age, genotype and retinopathy. J Fr Ophtalmol. 1997;20(9):653-8.
26. Downes SM, Hambleton IR, Chuang EL, Lois N, Serjeant GR, Bird AC. Incidence and natural history of proliferative sickle cell retinopathy: observations from a cohort study. Ophthalmology. 2005;112:1869-75.
27. Castro O. Management of sickle cell disease: Recent advances and controversies. Br J Hematol. 1999;107:2-11.
28. Sain MW, Oshinskie U, Greenberg BR. Case in point salmon patch-hemorrhages (nonproliferative retinopathy associated with sickle cell disease). Hosp Pract. 1997;32:239.
29. Kanski JJ. Oftalmología Clínica. Madrid: Elsevier. 2004. p. 480-3.
30. Hasan S, Elbedawi M, Castro O, Gladwin M, Palestine A. Central retinal vein occlusion in sickle cell disease. Br J Ophthalmol. 2003;87:342-7.
31. Clarke WN. Bilateral simultaneous retinal arteriolar obstruction in a child with hemoglobin SS sickle cell disease. J Am Ass Pediat Ophthalmol Strabismus. 2001;5:126-8.
32. Cao J, Mathers MK, Mc Leod DS. Angiogenic factors in human proliferative sickle cell retinopathy. Br J Ophthalmol. 1999;83:838-46.
33. Ariello LP. Clinical implications of vascular growth factors in proliferative retinopathies. Curr Opin Opthalmol. 1997;8:19-31.
34. Goldberg MF. Classification and pathogenesis of proliferative sickle retinopathy. Am J Ophthalmol. 1971;71:649-55.
35. Kent D, Arya R, Aclimandos WA, Bellingham AJ, Bird AC. Screening for ophthalmic manifestations of sickle cell disease in the United Kingdom. Eye. 1994;8:618-22.
36. Lutty GA, Goldberg MF. Ophtalmologic complications. En: Embury SH, Hebbel RP, Mohandas N, Steinberg MH, editors. Sickle cell disease. Basic principle and clinical practice. New York: Raven Press; 1994. p. 703-24.
37. National Institutes of Health. National Heart, Lung and Blood Institute. Division of Blood Diseases and Resources. The management of sickle cell disease. Bethesda: NIH Publication. 2002. p. 95-7.
Recibido: 12 de abril de 2008.
Aprobado: 2 de agosto de 2008.
Dra. Icilany Villares Álvarez. Servicio de Oftalmología. Hospital Universitario Dr. Gustavo Aldereguía Lima. Calle 51 A y Ave 5 de Septiembre. Cienfuegos. CP 55100. Cuba. E-mail: ivillares@gal.sld.cu


{kind=link}