










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Oftalmología
versión On-line ISSN 1561-3070
Rev Cubana Oftalmol vol.27 no.4 Ciudad de la Habana oct.-dic. 2014
INVESTIGACIÓN
Estudio generacional en familias con distrofia corneal endotelial de Fuchs
Generational study conducted in families with Fuchs' endotelial corneal dystrophy
Dra. Madelaine López González,I Dr. Urbano Rodríguez de la Paz,II Dra. Silvia María López Hernández,II Dra. Damarys García Gómez,III Dra. Suzel Lapido Polanco,II Dr. Waldemar Baldoquin RodríguezIV
I Hospital Pediátrico Docente "Juan Manuel Márquez". La Habana, Cuba.
II Instituto Cubano de Oftalmología "Ramón Pando Ferrer". La Habana, Cuba.
III Centro Nacional de Genética Médica. La Habana, Cuba.
IV Servicios Médicos de Cuba. La Habana, Cuba.
RESUMEN
Objetivos: identificar la presencia de rasgos clínicos de distrofia corneal endotelial de Fuchs en familiares de pacientes con este diagnóstico.
Métodos: se realizó un estudio observacional, descriptivo y transversal de 15 familias trigeneracionales de pacientes con distrofia corneal endotelial de Fuchs, captados en consulta de córnea del Instituto cubano de Oftalmología "Ramón Pando Ferrer" del año 2008 al 2011. La muestra quedó constituida por un total de 78 pacientes. Se utilizaron planillas de recolección de datos para estudiar las siguientes variables de interés: gutas corneales según grados, clasificación clínica, edad, sexo y grado de parentesco según árbol genealógico familiar. Se resumieron las variables cualitativas utilizando frecuencias absolutas y relativas porcentuales.
Resultados: del total de descendientes directos, el 44,1 % presentó gutas en su córnea. El 34,6 % de los pacientes estudiados presentaron la enfermedad y el 23,1 % córnea guttata. La distrofia y la córnea guttata se constataron más en el sexo femenino, con mayor incidencia en mayores de 60 años para los primeros. Los descendientes del primer nivel evidenciaron más afectación corneal que los de segundo nivel, con el 75,9 y el 23,5 % respectivamente. Todas las familias presentaron miembros afectados en su descendencia.
Conclusiones: la mayor parte de las familias de pacientes con distrofia corneal endotelial de Fuchs presentan cambios endoteliales en las córneas, por lo que estas deben ser incluidas en el estudio con fines diagnósticos y preventivos.
Palabras clave: distrofia endotelial de Fuchs, distrofia corneal, herencia, estudio familiar.
ABSTRACT
Objective: to identify clinical features of Fuchs' endothelial corneal dystrophy in relatives of patients with this diagnosis.
Methods: bservational, descriptive and transversal study of 15 three- generation families of those patients with Fuchs' endothelial corneal dystrophy who went to the cornea consultation of the "Ramón Pando Ferrer", Cuban Institute of Ophthalmology from 2008 to 2011. The sample was made up of 78 patients. Data collection forms were used to study the following variables of interest: Cornea Guttata by degree, clinical classification, age, sex and degree of relationship as family tree. Qualitative variables were summarized using percentage absolute and relative frequencies.
Results: of the total number of direct descendants, 44,1 % had cornea guttata; 34,6 % of the patients presented with Fuchs' endothelial corneal dystrophy and 23,1 % of them had cornea guttata. Dystrophy and cornea guttata were found more frequently in females, with the highest incidence of the former in patients over 60 years. First degree descendants showed more corneal involvement than the second degree ones (75,9 and 23,5 %, respectively). All the families had offspring affected by the disease.
Conclusions: most of the members of the families suffered corneal endothelial changes, so they should be included in the study for diagnostic and preventive purposes.
Key words: Fuchs' endothelial corneal dystrophy, corneal dystrophy, genetic inheritance, family study.
INTRODUCCIÓN
La denominación "distrofia corneal" (DC) fue utilizada por vez primera por Ernst Fuchs a principios del siglo XX, quien la tomó del griego dys (alteración) y trophé (nutrición). Hoy en día se conoce que las distrofias no guardan relación con la nutrición, pero el término se ha mantenido a pesar de conocer que su desarrollo progresivo se relaciona con cambios patológicos como resultado de defectos genéticos.1-3
La córnea, estructura donde aparece este tipo de afectación, es la porción anterior y transparente de la cubierta externa del ojo, formada por cinco capas: epitelio, capa de Bowman, estroma, membrana de Descemet (MD) y endotelio.1,3,4
Para una mejor comprensión y estudio de las distrofias se determinó darles un orden o clasificación, y quedaron divididas en anteriores (epiteliales y subepiteliales), estromales y posteriores (endoteliales). La distrofia corneal endotelial de Fuchs (DCEF) está comprendida dentro de las distrofias posteriores. Se trata de un trastorno degenerativo específico y progresivo del endotelio corneal que afecta aproximadamente el 4 % de la población mayor de 40 años, y mucho más a mujeres que a hombres.1
En la DCEF la membrana de Descemet puede estar considerablemente afectada con engrosamiento y excrecencias posteriores denominadas clínicamente gutas o verrugas, las cuales se catalogan como pseudogutas cuando se relacionan con historia de traumatismo, cirugía o inflamación intraocular. Es la causa más común de disfunción endotelial endógena descrita hace más de 100 años.1,5
Hace más de medio siglo que se conocen formas familiares de DCEF, que exhiben un patrón hereditario autosómico dominante en más del 50 % de los casos.1,6-8 Lo cierto es que aunque la mayoría de los estudios señalan esto, muchos casos clínicos de DCEF parecen ser esporádicos, que conjuntamente con la preponderancia femenina fueron explicados por una penetración incompleta, aunque se ha estudiado que posiblemente la penetración sea de un 100 % con una expresividad variable.1, 6
Con el desarrollo actual de la Oftalmología en Cuba y la frecuente realización de trasplantes corneales es imprescindible tener en cuenta la morfología de la córnea del donante, ya que estudios realizados han reportado que luego de cierto tiempo comienzan a presentar cambios endoteliales característicos de la distrofia corneal endotelial de Fuchs (DCEF), que da al traste con el éxito de la intervención, por lo que se presume sea por la presencia de esta enfermedad en la córnea donante.1,9
Teniendo en cuenta lo anteriormente planteado, podríamos inferir la importancia que tiene el conocimiento por parte del cirujano y del paciente sobre el riesgo aumentado de fallo endotelial en caso de practicarse una cirugía del segmento anterior en aquellos que presenten esta afección. De igual manera, sería preciso examinar a los familiares de los pacientes con distrofia aunque no presenten síntomas de la enfermedad con el objetivo de identificar la presencia o no de esta en etapas tempranas, lo que sería una herramienta de gran valor predictivo en manos del especialista en oftalmología, que le permitiría dar una adecuada y especial atención a pacientes y familiares.
Con este trabajo se pretende identificar la presencia de rasgos clínicos de distrofia corneal endotelial de Fuchs en familiares de pacientes con este diagnóstico.
MÉTODOS
Se realizó un estudio observacional, descriptivo y transversal de 15 familias de tres generaciones cada una, con al menos un miembro con historia conocida de DCEF captado en consulta de córnea del Instituto Cubano de Oftalmología "Ramón Pando Ferrer", del año 2008 al 2011. Cada familia se constituyó a partir de 15 pacientes con diagnóstico previo de DCEF y sus 63 descendientes directos, que sumaron un total de 78 miembros.
Se tomaron en cuenta como criterios de inclusión: que tuvieran tres generaciones vivas partiendo del paciente de forma ascendente o descendente; más de 10 años de edad; pacientes sin antecedentes oftalmológicos previos de traumas e inflamaciones oculares; usuarios de lentes de contacto o de intervención quirúrgica del segmento anterior y que aceptaran participar en el estudio.
Para la investigación se tomaron los datos de las planillas de recolección y de la base de datos del equipo de Microscopio Confocal (Confoscan 4 NIDEK Technology), donde se le realizó la microscopia confocal (MC) a cada paciente y familiar.
Para dar salida a los objetivos del estudio se operacionalizaron las variables descritas a continuación:
- Gutas corneales (cualitativa ordinal) según grados (clasificación de Krachmer):10
-Edad (cuantitativa continua) y sexo (dicotómica).
Se le confeccionó el árbol genealógico a cada familia, lo que permitió conocer el grado de parentesco y de asociación con la enfermedad de cada uno de sus integrantes, considerando:
-Descendientes de primer nivel: representados por los hijos de los 15 pacientes con DCEF que fueron captados en consulta.
-Descendientes de segundo nivel: representados por los nietos de los 15 pacientes con DCEF que fueron captados en consulta o lo que es lo mismo, los hijos de los descendientes de primer nivel.
A todos los miembros de las familias que cumplían con los criterios de inclusión, independientemente del grado de parentesco, se les realizó interrogatorio y examen oftalmológico en LH (BQ 900 Zeiss) en busca de las variables de interés en el estudio.
Los familiares que no eran descendientes directos de los pacientes con DCEF y que en consulta bajo observación biomicroscópica no presentaron gutas corneales, no fueron incluidos en el análisis de los resultados del estudio (aunque se les realizó el resto de los exámenes como a los demás familiares), ya que la ausencia de estas excrecencias descartó la presencia de DCEF y, por tanto, la posibilidad de transmisión de sus características a los descendientes por parte del otro progenitor sin relación directa con las familias estudiadas.
Los datos recogidos en las planillas de recolección se pasaron a una base de datos (Excel), para su procesamiento estadístico. Se utilizaron en general los métodos de la estadística descriptiva. Se resumieron las variables cualitativas utilizando frecuencias absolutas y relativas porcentuales.
Se les informó a pacientes y familiares sobre los elementos de juicio médico y éticos que sustentan esta investigación, brindándoles la posibilidad de retirarse en cualquier momento del estudio. Tanto los incluidos en el estudio como los no incluidos recibieron los beneficios del servicio indistintamente.
RESULTADOS
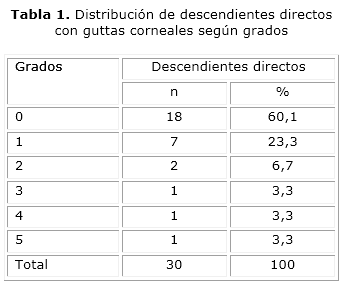
En las 15 familias estudiadas se observaron 63 descendientes directos; de ellos hubo un total de 30 miembros afectados, para un 44,1 %. En la tabla 1 se encuentran distribuidos todos los descendientes que presentaron cambios biomicroscópicos corneales tipo gutas. Los pacientes se concentraron en mayor número entre el grado 0 y el grado 1.
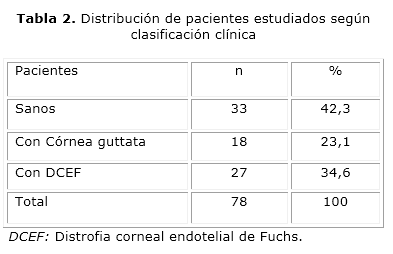
Según la clasificación clínica realizada (tabla 2), del total de pacientes estudiados predominaron los individuos con algún tipo de lesión endotelial (57,7 %), con DCEF (34,6 %) y con córnea guttata (23,1 %).
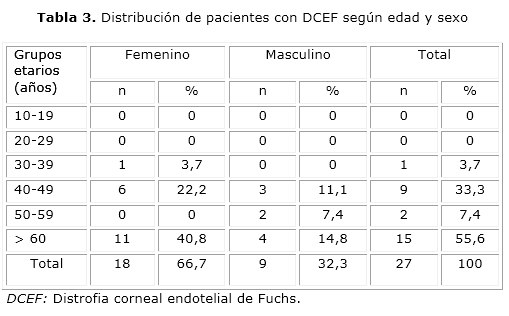
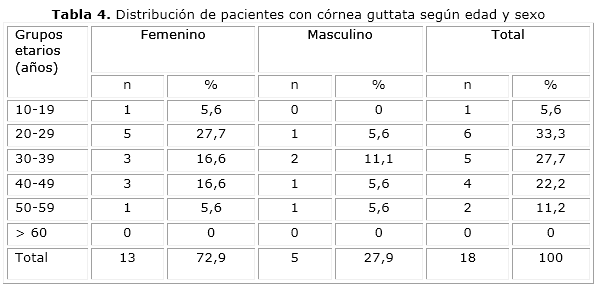
En los casos con DCEF y córnea guttata predominó el sexo femenino, con 66,7 y 72,9 % respectivamente. En ambos casos el sexo femenino duplicó en frecuencia al masculino; sin embargo, el grupo de edades más afectado, con 55,6 %, fue el de mayores de 60 años para los pacientes con distrofia y el de 20 a 29 años para los que tenían córnea gutatta, con el 33,3 % (tablas 3 y 4).
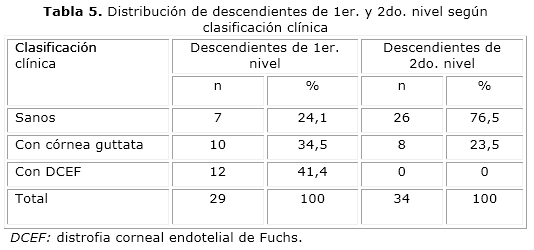
Los pacientes descendientes de 1er. y 2do. nivel fueron distribuidos según la clasificación clínica en la tabla 5, donde se evidenció que la mayoría de los pacientes con alguna afectación corneal se encontraban entre los descendientes de 1er. nivel, con 22 casos, para un 75,9 %. Nótese que la DCEF solo se observó en descendientes de este nivel, mientras que en los descendientes de 2do. nivel solo 8 pacientes presentaron cambios corneales, para el 23,5 %.

DISCUSIÓN
En sus inicios la DCEF debuta con una serie de excrecencias centrales corneales en la membrana de Descemet, también denominadas guttas; a medida que la enfermedad avanza aparece mayor disfunción del endotelio corneal, que sintomáticamente se traducen en una disminución de la agudeza visual por la aparición de edema corneal, que puede llegar a ser tan severa que lleva secundariamente a muchos de los pacientes a la realización de un trasplante corneal en busca de una mejoría clínica.1,11-15
En este estudio, dentro de los familiares sin diagnóstico previo de DCEF y que presentaron gutas corneales en el examen biomicroscópico y al MC, fueron diagnosticados nuevos casos con la enfermedad corneal y otros con córnea guttata, lo que está relacionado con la existencia de formas familiares de ambas afecciones.1,14-17
Moschifar describió la presencia de córnea guttata en familiares de pacientes con DCEF que iban a ser sometidos a procedimientos de cirugía refractiva.18 Según los datos que fueron obtenidos y analizados en la investigación, más de la mitad de los miembros de las familias estudiadas presentaron algún tipo de alteración endotelial, ya sea por córnea guttata o por la presencia de DCEF.
El sexo, predominantemente el femenino, es clásico en este tipo de afección y no se comportó de manera diferente en este estudio, que mostró una relación 2: 1. Muchos estudios describen resultados similares, refiriendo proporciones que van desde 2-3: 1, los más frecuentes, hasta 4: 1.1,2,16,19
Los grupos de edades entre 10 y 29 años no presentaron pacientes con DCEF; estos se distribuyeron en su mayoría en edades posteriores a los 40 años de edad, lo que está en correspondencia con la edad de debut de esta enfermedad. Si tenemos en cuenta que todos los pacientes de los que partió nuestro estudio sobrepasaban los 60 años, es lógico encontrar el mayor número de enfermos superior a este rango de edad, de lo que se deriva que los pacientes que están representados en el grupo de 40-49 años se corresponden con los nuevos pacientes diagnosticados en quienes se buscó la presencia de la enfermedad.11,20
En la bibliografía consultada se plantea que la frecuencia de la DCEF en pacientes mayores de 40 años va desde el 10 % hasta el 70,4 %, aunque es descrita con mayor frecuencia en edades posteriores a la sexta y séptima década de la vida.2,17
Contrario a la distribución de pacientes con DCEF, los descendientes directos que presentaron córnea guttata se encontraron en edades anteriores a la quinta década de la vida, lo que resulta ser un dato de gran interés si tenemos en cuenta que se trata de pacientes con antecedentes importantes de familiares con DCEF y que además presentan en sus córneas cambios endoteliales que, aunque no clasifican como enfermos por el momento, no deja de constituir un riesgo quizás predictor de posible desarrollo de la enfermedad en edades posteriores, o un debut anticipado de esta al ponerse en contacto con algún factor que la descompense.
Estudios recientes han probado la presencia de mutaciones en el gen para la cadena a2 (COL8A2) en los brazos cortos del cromosoma 1, que muestra una histopatología atípica con debut de la enfermedad en etapas tempranas o infantiles, por lo que ya se maneja en la literatura los términos DCEF de comienzo temprano para referirse a esta enfermedad que lleva implícito las mutaciones genéticas y DCEF de inicio tardío cuando se trata de aquella que se presenta luego de los 40 años de edad.19,21
En la investigación, más de la mitad de los pacientes descendientes de primer nivel presentaron algún tipo de afectación corneal, ya sea por la presencia de distrofia de Fuchs o de córnea guttata, mientras que en los de segundo nivel los sanos representaron las tres cuartas partes de sus miembros, lo que está relacionado con el tipo de patrón de herencia seguido por esta enfermedad en las familias estudiadas, correspondiente al tipo autosómico dominante (AD), donde los rasgos son heredados de padres a hijos.6 En la segunda descendencia no se ven individuos afectados con la enfermedad, por la edad de debut de esta, y son muy jóvenes los miembros que componen este grupo, por lo que es más probable encontrar córnea guttata en esta generación.2,11-16,20,22
La herencia autosómica dominante ha sido ampliamente descrita por muchos autores; sin embargo, otros plantean un porciento no despreciable de la enfermedad que se muestra de manera esporádica.1 Las dos terceras partes de las familias estudiadas presentaron miembros con DCEF, lo que ratifica lo antes expuesto sobre el patrón de herencia seguido por esta enfermedad, donde se supone encontrar descendientes con similares alteraciones del gen que la codifica, aunque en este caso estudiamos solamente la expresión fenotípica de la distrofia. Krachmer describió un total de hasta 20 familiares afectados con DCEF en una familia extensa al hacer un estudio a familias con este tipo de afección.10
La córnea guttata fue vista en el primer y segundo nivel de descendencia en más de la mitad de las familias estudiadas, lo que demuestra la correspondencia de esta afectación corneal con un patrón autosómico dominante, por la transmisión vertical de los rasgos, y se vieron afectadas todas las generaciones, con un comportamiento similar al seguido por la distrofia de Fuchs en nuestro estudio.11,16,20,21 Aunque se han descrito formas familiares de córnea guttata, casi siempre su descubrimiento ha sido casual al hacer el estudio en familias con DCEF.18
Los estudios genéticos tienden por lo general a ser muy costosos, además de la implicación del tiempo y la dedicación a estos. La mayoría de los autores escogen para sus estudios muy pocas familias, casi siempre una o dos, aunque estas sean numerosas, y se les realiza el estudio de sus genes luego de detectar la presencia de afección corneal en consulta. El Doctor Olof describe a una familia donde más de la mitad de sus integrantes presentaron DCEF luego de estudios genéticos que les fueron realizados.22
En la escasa bibliografía existente sobre estudios familiares de este tipo, el compromiso fenotípico de las familias no es detallado; casi siempre se busca directamente la presencia de la enfermedad clínica para la realización de estudios de sus genes y no de su expresión. Las demás manifestaciones de cambios endoteliales quizás precursoras de la enfermedad, como es el caso de la córnea guttata, si llega a ser mencionada casi siempre es reportada como hallazgo casual en otro tipo de investigación.
La DCEF es una enfermedad ocular con una evolución tórpida e invalidante para el individuo que la presenta, y provoca un déficit visual paulatino que la mayoría de las ocasiones termina en un transplante corneal para lograr mejor visión. Este es un tratamiento definitivo y costoso, que en ocasiones también falla. Existen muy pocos estudios que abarcan este tema. Profundizar en su conocimiento en búsqueda de posibilidades futuras de predecir o no la presencia y desarrollo de esta enfermedad en familiares de pacientes que ya se les ha diagnosticado, tendría gran impacto científico y social.
REFERENCIAS BIBLIOGRÁFICAS
1. Barraquer RI, De Toledo MC, Torres E. Distrofias y degeneraciones corneales. Atlas y Texto. Barcelona: Expas; 2004. p. 20921.
2. Rosenblum P, Stark WJ, Maumenee IH, Hirst LW, Maumenee AE. Hereditary Fuchs' dystrophy. Am J Ophthalmol. 1990;90:455-62.
3. Chang SW, Tuli S, Azar D. Corneal Dystrophies. Genetic diseases of the eye: a textbook and atlas. New York: Oxford University; 1998.
4. Urquhart JE, Biswas S, Black GC, Munier FL, Sutphin J. Exclution of COL8A1, the gen encoding the (VIII)2 chain of type VIII collagen, as a candidate for Fuchs endothelial dystrophy and posterior polymorphous corneal dystrophy. Brit J Ophthalmol. 2006;90:1430-31.
5. Ula VJ, Rawe I, Bitar MS, Cheng Z, Harris DL, Colby K, Joyce NC. Decreased Expression of Peroxiredoxins in Fuchs' Endothelial Dystrophy. Invest Ophthalmol. Vis Sci. 2008;49(7):2956-63.
6. Biswas S, Munier FL, Yardley J. Missense mutation in COL8A2, the gene encoding the a2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum Mol Genet. 2006;10:2415-23.
7. Aldave AJ, Rayner Sa, Salem AK. No pathogenic mutations identified in the COL8A2 genes in familial with Fuchs' corneal dystrophy. Invest Ophthalmol Vis Sci. 2006;47(9):3787-90.
8. Mok JW, Kim HS, Joo CK. Q455V mutation in COL8A2 is associated with Fuchs' corneal dystrophy in Korean patients. Eye. 2009;23:895-903.
9. Okumura N, Koizumi N, Ueno M, et al. Enhancement of corneal endothelium wound healing by Rho-associated kinase (ROCK) inhibitor eye drops. Br J Ophthalmol. 2011;95:1006-9.
10. Krachmer JH, Purcell JJ, Young CW, Bucher KD. Corneal endothelial dystrophy. A study of 64 families. Arch Ophthalmol. 1978;96:2036-9.
11. Lorenzetti DWC, Uotila MH, Parikh N, Kaufman HE. Central cornea guttata. Incidente in the general population. Am J Ophathalmol. 1967;64:115558.
12. Roth SI, Stock EL, Justabha R. Endothelial inclusions in Fuchs' corneal dystrophy. Hum Pathol. 2006;18(4):338-41.
13. Patel SV, Baratz KH, Hodge DO, et al. The effect of corneal light scatter on vision after Descemet stripping with endothelial keratoplasty. Arch Ophthalmol. 2009;127:153-60.
14. Veitía RZ, Bauza FY, Hernández SR, Ramos LM, Curbelo CL, López HI. Estudio comparativo de la pérdida celular endotelial entre las técnicas de facoemulsificación por ultrachop y prechop. Rev Cub Oftalmol. 2010 [citado 20 de agosto de 2014];23(Supl.2). Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-21762010000400007&lng=es&nrm=iso&tlng=es
15. Hecker AL, McLaren JW, Bachman LA, Sanjay V, Patel BA. Anterior Keratocyte Depletion in Fuchs' Endothelial Dystrophy. Arch Ophthalmol. 2011;129(5):555-61.
16. Cross HE, Maumenee AE, Cantolino SJ. Inheritance of Fuchs` endothelial dystrophy. Arch Ophthalmol. 1991;85:268-72.
17. Zoega GM, Fujisawa A, Sasaki H, et al. Prevalence and risk factors for cornea guttata in the Reykjavik Eye Study. Ophthalmology. 2006;113:565-9.
18. Moshirfar M, Feiz V, Feilmeier MR, Kang PC. Laser in situ keratomileusis in patients with corneal guttata and family history of Fuchs' endothelial dystrophy. J Cataract Refract Surg. 2005;31(12):2281-6.
19. Bolaños G. Distrofia endotelial de Fuchs. OSL, St. Luc¡a: Oftalmol. 2009;8(3):85-94.
20. Bergmanson JP, Sheldon TM, Goosey JD. Fuchs' endothelial dystrophy: a fresh look at an aging disease. Ophthalmic Physiol Opt. 1999;19:210-22.
21. Gottsch JD, Eghrari OA. Fuchs' corneal dystrophy. Cataract, Cornea and External Disease Service. Wilmer Eye Institute. Expert Rev Ophthalmol. 2010;5(2):147-59.
22. Sundin OH, Jun AS, Broman KW, Liu SH, Sheehan SE, Vito EC, et al. Linkage of lateonset Fuchs corneal dystrophy to a novel locus at 13 ptel13 q12.13. Invest Ophthalmol Vis Sci. 2006;47:140-5.
Recibido: 20 de junio de 2014.
Aprobado: 3 de julio de 2014.
Dra. Madelaine López González. Hospital Pediátrico Docente "Juan Manuel Márquez". Marianao, La Habana, Cuba. Correo electrónico: madelopezg@infomed.sld.cu

