










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Oftalmología
versión On-line ISSN 1561-3070
Rev Cubana Oftalmol vol.31 no.1 Ciudad de la Habana ene.-mar. 2018
PRESENTACIÓN DE CASO
Retinoblastoma: una presentación tardía y atípica
Retinoblastoma: late-onset and atypical presentation
Liudmira González Rodríguez,I Mercedes Cárdenas Bruno,I Myrna I. Moreno Miravalles,I Lázaro Vigoa Aranguren,II Ernesto Alemañy RubioII
I Hospital Pediátrico "Juan Manuel Márquez". La Habana, Cuba.
II Instituto Cubano de Oftalmología "Ramón Pando Ferrer". La Habana, Cuba.
RESUMEN
El retinoblastoma es el tumor intraocular maligno más frecuente en la niñez y representa alrededor del 4 % de todos los cánceres de esta etapa de la vida y menos del 1 % en general. Este reporte describe la presentación tardía de retinoblastoma en forma de panuveítis en un adolescente de 11 años, diagnosticado inicialmente como toxocariasis ocular. Esta enfermedad es rara en pacientes mayores de 8 años, pero tiene que ser incluida como diagnóstico diferencial. El diagnóstico tardío o erróneo constituye un riesgo para la vida y peor pronóstico visual.
Palabras clave: retinoblastoma; presentación tardía; uveítis; diagnóstico diferencial.
ABSTRACT
Retinoblastoma is the most common malignant intraocular tumor in childhood, representing about 4% of all cancers at that stage of life and 1% in general. A description is provided of a late-onset panuveitic retinoblastoma in an 11-year-old male patient initially diagnosed with ocular toxocariasis. The disease is rare in people over 8 years of age, but it should be included in the differential diagnosis. Late or mistaken diagnosis constitutes a risk to life and a worse visual prognosis.
Key words: retinoblastoma; late onset; uveitis; differential diagnosis.
INTRODUCCIÓN
El retinoblastoma es el tumor intraocular maligno más frecuente en la niñez y representa alrededor del 4 % de todos los cánceres de esta etapa de la vida y menos del 1 % en general.1-3 El 90 % de los pacientes son diagnosticados con menos de 3 años de edad y el resto se diagnostica, en la mayoría de los casos, antes de los 5 años.4 Es muy raro su diagnóstico en pacientes mayores de 8 años.5 Aunque su incidencia es baja (1 caso entre 15 000 y 20 000 nacidos vivos). Su posibilidad diagnóstica, junto a su frecuente carácter hereditario (en un 40-50 % de los casos se transmite con carácter autosómico dominante con alta penetrancia), ha hecho del retinoblastoma un paradigma del cáncer hereditario.6,7
Además de su aporte al mejor entendimiento de la genética del cáncer, el retinoblastoma es un ejemplo interesante de la disparidad entre los países desarrollados y los que están en vías de desarrollo. En los primeros, la tasa de supervivencia de los pacientes es del 95 %, lo cual es atribuible a la accesibilidad a los servicios de salud para el diagnóstico y el tratamiento precoz de la enfermedad. Como resultado, salvar el globo ocular y la posibilidad de visión útil se han convertido en los principales objetivos en esos países. Sin embargo, en los países en vías de desarrollo, el retinoblastoma es aún una enfermedad que provoca la muerte de muchos niños,8-10 con una mortalidad del 65 % en Nigeria y del 95 % en las Filipinas.11 Si no es tratado, este tumor provoca invasión local de los tejidos y metástasis a distancia, lo que causa la muerte mayormente dentro de los dos años de su debut.12
Cuba, a pesar de ser un país subdesarrollado, presenta tasas de supervivencia parecidas o iguales a los países del primer mundo; pero todavía existen errores diagnósticos que retrasan el tratamiento adecuado de estos pacientes. Presentamos un caso de retinoblastoma en Cuba para describir la presentación tardía y atípica de la enfermedad y la importancia de un adecuado diagnóstico diferencial.
PRESENTACIÓN DEL CASO
Paciente masculino de 11 años de edad, atendido en el Instituto Cubano de Oftalmología "Ramón Pando Ferrer" en enero del año 2017. Comenzó con ojo rojo y dolor ocular en el ojo derecho (OD) 6 meses antes y fue diagnosticado y tratado por toxocariasis ocular en el hospital de su área de salud. Pero, por el empeoramiento del cuadro clínico, a pesar del tratamiento recibido y del largo período de evolución, fue remitido a nuestro Instituto para una reevaluación.
Al interrogatorio realizado en el momento de la admisión, el paciente y sus familiares refirieron antecedentes generales y oculares de salud anterior, negaron antecedentes patológicos familiares oculares y los factores de riesgo para adquirir enfermedades infectocontagiosas. Al examen oftalmológico la mejor agudeza visual corregida fue de 0,2 en el ojo derecho y de 1,0 en ojo izquierdo (OI). La presión intraocular (PIO) fue de 21 mmHg en el OD y de 19 mmHg en el ojo izquierdo. Al examen del OD presentaba: inyección cilioconjuntival moderada, hipopion de células grandes y granulosas, asociado a una capa de sangre, que cubrían el iris inferior y parte del área pupilar; turbidez del humor acuoso (flare) 2+; celularidad del humor acuoso 4+; neovascularización del iris en toda la superficie visible de este; sinequias posteriores en sector superior y opacidad cortical sectorial del cristalino.
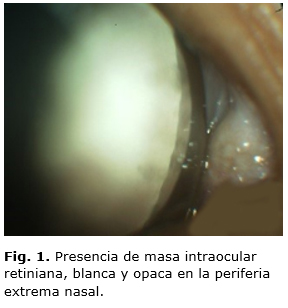
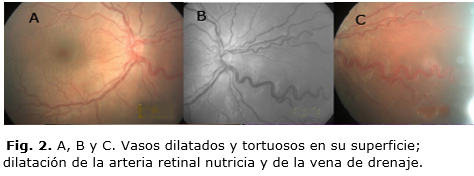
Al examen del fondo de ojo dilatado se observó presencia de masa intraocular retiniana, blanca y opaca en la periferia extrema nasal que cabalgaba la ora serrata (Fig. 1), con vasos dilatados y tortuosos en su superficie y dilatación de la arteria retinal nutricia y de la vena de drenaje (Fig. 2). Además, opacidades vítreas pequeñas y blancas, algunas desprendidas de la masa (siembras). En el OI, tanto el segmento anterior, como el posterior, eran normales.
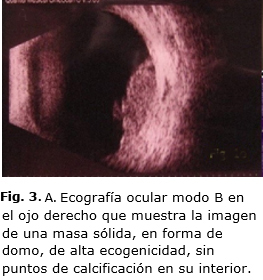
Se le realizó una ecografía ocular modo B (Quantel Medical CineScan. S V.5.05) en el ojo derecho que mostró la imagen de una masa sólida, en forma de domo, de alta ecogenicidad, sin puntos de calcificación en su interior y dimensiones de 16 x 10 x 8 mm. La retina se encontraba aplicada (Fig. 3).
Se realizó retinografía e imágenes del segmento anterior a color, con cámara midriática [FF- 450 plus IR. Carl Zeiss ®, Meditec AG. 07740 Jena, Alemania (2005). Megaplus Camera. Modelo 1.6i/10BIT (Jencam)], la cual no permite tomar imágenes de más de 50 grados y en localizaciones anteriores al ecuador. El uso de este medio fue necesario por no disponer de cámara de fondo de campo ultra-amplio, ni cámara de fondo pediátrica (RetCam), que permiten adquirir imágenes hasta de 150 grados.
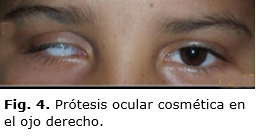
Se consideró como diagnóstico un retinoblastoma endofítico unilateral, del grupo V por la clasificación de Reese-Ellsworth, y del grupo E por la Clasificación Internacional del Retinoblastoma (ICRB, por sus siglas en inglés). Se sugirió la enucleación del globo ocular derecho y el implante orbitario integrado de hidroxiapatita porosa HAP- 200, Coralina ®,Neuronic, tipo IO 18, de acuerdo con los cálculos realizados. Se colocó un conformador en el transquirúrgico y a las seis semanas una prótesis ocular cosmética (Fig. 4).
Después de realizada la enucleación, el informe anátomo-patológico describió macroscópicamente un globo ocular con diámetro anteroposterior de 21,79 mm, diámetro transversal y vertical de 11 mm y segmento de nervio óptico de 8 mm. Al corte se observó notable masa tumoral blanco-amarillenta, extendida por el sector nasal posecuatorial hasta la ora serrata, e invasión de la cámara anterior.
Microscópicamente mostró un retinoblastoma poco diferenciado con extensas áreas de necrosis intratumoral, infiltración mínima de la coroides y nidos de células tumorales en cámara anterior. No infiltración de esclera, ni nervio óptico. El diagnóstico histopatológico definitivo fue: retinoblastoma poco diferenciado del ojo derecho (Fig. 5). El estadiamento oncológico fue T3c, N0 y M0. No fue necesario el tratamiento con quimioterapia sistémica, según protocolo.

DISCUSIÓN
El retinoblastoma se presenta generalmente en pacientes menores de 4 años, con una edad media al diagnóstico de 18 meses. Para la forma unilateral es cerca de los 24 meses, y antes de los 12 meses en los casos bilaterales. Su diagnóstico en mayores de 8 años es raro. De acuerdo con lo planteado por Shields y otros, solo el 8,5 % de los pacientes con retinoblastoma tienen más de 5 años en el momento del diagnóstico y todos los reportes de estos casos son esporádicos.13
Con el desarrollo alcanzado en el diagnóstico y las modalidades de tratamiento, el diagnóstico precoz y el tratamiento rápido y oportuno han mejorado considerablemente la superviviencia global, la ocular y la visión de los pacientes con esta enfermedad.14 De lo contrario, este tumor intraocular crece rápidamente y puede ser fatal.15
El paciente presentó los síntomas iniciales a la edad de 11 años, pero no se puede decir que el tumor estuvo creciendo lentamente desde la infancia temprana; sino que la explicación más probable, según algunos investigadores,16 es la reactivación de un retinocitoma ya existente en la retina periférica, nunca diagnosticado, por la ausencia de antecedentes de un examen oftalmológico previo.
Gallie y otros sugieren una modificación en el modelo original de mutación de la oncogénesis propuesto por Knudson para explicar la presencia de un retinocitoma y la baja penetrancia de una mutación del gen Rb1 en algunas familias. Es bien aceptado que el retinoblastoma hereditario con una mutación germinal se desarrollará si un segundo evento (mutación) ocurre en retinoblastos inmaduros susceptibles (primitivos); mientras que el retinocitoma será si el segundo evento ocurre en retinoblastos maduros. Otra de las explicaciones planteadas para la presentación tardía del retinoblastoma es la persistencia de células retinales embrionarias raras que pueden transformarse en malignas en etapas tardías de la vida.17-19
Por otra parte, el retinoblastoma puede desarrollarse en ojos con alteraciones prexistentes que dificulten su diagnóstico, como catarata, coloboma, otras malformaciones coriorretinianas, entre otras, y esto puede ser la causa en ocasiones de un diagnóstico tardío; 20 pero el paciente que se presenta en este reporte no tiene antecedentes patológicos personales oculares anteriores.
La leucocoria es el signo clínico más común en el retinoblastoma, la cual, al principio, puede ser intermitente de acuerdo con la localización del tumor. Sin embargo, se debe considerar como un síntoma tardío. Es seguida en frecuencia por el estrabismo que al inicio también puede ser intermitente. Con menos frecuencia, consideradas formas atípicas de presentación están la endoftalmitis, la uveítis, la heterocromía y el glaucoma agudo. Estas podrían enmascarar el diagnóstico final. En el caso de la uveítis no debe pasar inadvertida, y al observarla debemos agudizar nuestra habilidad clínica para sospechar entidades comunes, pero las que son poco frecuentes también. Así, enfermedades de origen infeccioso o inflamatorio pueden dar cuadros clínicos similares a los de un retinoblastoma. Los casos muy avanzados pueden presentarse con proptosis, celulitis orbitaria, hipertensión endocraneana por metástasis en el SNC, entre otras.21
Estudios retrospectivos realizados en países subdesarrollados muestran que la duración de los síntomas oculares oscila entre días y meses antes de que surja la sospecha de un tumor, y el paciente sea entonces remitido a un servicio de oncología ocular. Es posible, aún en hospitales especializados, que el diagnóstico pase inadvertido por algún tiempo y los pacientes, al llegar al centro de referencia para descartar un tumor intraocular, puedan tener ya varios meses de estar recibiendo un tratamiento inadecuado.
Los principales diagnósticos diferenciales son la persistencia del vítreo primario hiperplásico 28 %, y la enfermedad de Coats 26 %; menos común la uveítis por Toxocaras 16 %. El diagnóstico diferencial del retinoblastoma depende principalmente de la forma de presentación. En el caso específico de la uveítis, este es un signo clínico que se asocia a diversas enfermedades, dentro de las cuales se pueden mencionar la toxocariosis ocular y la toxoplasmosis.
Cuando se presenta como una leucocoria se deben tener en cuenta también otras entidades como: enfermedad de Coats, persistencia de la vasculatura fetal, catarata congénita y retinopatía de la prematuridad. Los antecedentes perinatales normales descartarán la retinopatía de la prematuridad. El examen con la lámpara de hendidura revelará la transparencia del cristalino. Las características de la lesión tumoral en el ultrasonido ocular y en la resonancia magnética no se correlacionarán con una enfermedad de Coats o con la persistencia de la vasculatura fetal. Más bien, todos los signos encontrados al examen físico y las imágenes nos guiarán hacia el diagnóstico certero de retinoblastoma.
El papel del oftalmólogo en estos casos es llegar a una impresión diagnóstica adecuada cuando los datos nos dan suficiente evidencia de que puede estar implicada una entidad maligna. Para poder hacer el diagnóstico preciso, debemos empezar por examinar detalladamente al paciente; tener conocimiento de los diagnósticos diferenciales; emplear correctamente los exámenes auxiliares y analizarlos objetivamente.22-23
Nos queda la enseñanza de que la sospecha clínica se puede lograr recordando dos aspectos importantes: el conocimiento de los diagnósticos diferenciales y el examen repetido de pacientes que no mejoran a pesar del tratamiento médico adecuado. El diagnóstico tardío o erróneo contribuye a un mayor riesgo para la vida del paciente y a un peor pronóstico visual. La optimización de estrategias de educación en los signos tempranos del retinoblastoma y de diagnóstico precoz pueden jugar un papel importante en la salud ocular.
Conflicto de intereses
Los autores declaran que no existe conflicto de intereses en el presente artículo.
REFERENCIAS BIBLIOGRÁFICAS
1. Gelaw Y, Shoukry SM, Othaman IS. Unusually very late- onset new growth of intraocular retinoblastoma: a case report and review of literature. Am J Ophthalmol Case Reports. 2017 [citado 10 de enero de 2018]. Disponible en: http://dx.doi.org/10.1016/j.ajoc.2016.12.013
2. Chintagumpala M, Chévez-Barrios P, Paysse EA, Plon SE, Hurwitz R. Retinoblastoma: review of current management. Oncologist. 2007;12:1237-46.
3. Othman IS. Retinoblastoma major review with updates on Middle East management protocols. Saudi J Ophthalmol. 2012;26:163-75.
4. Abramson DH. Retinoblastoma in the 20th Century. Invest Ophthalmol Vis Sci. 2005;46(8):2684-91.
5. Sharifzadeh M, Ghassemi F, Amoli FA, Rahmanikhah E, Tabatabaie SZ. Retinoblastoma in Adults: a case report and literature review. J Ophthalmic Vis Res. 2014;9(3):388‑91.
6. Vogt F. Genetics of retinoblastoma. Hum Genet. 1979;52:1-54.
7. Alonso J, Palacios I, Gámez A, Camino I, Frayle H, Menéndez I et al. Diagnóstico molecular del retinoblastoma: epidemiología molecular y consejo genético. Med Clin (Barc). 2006;126(11):401-5.
8. Chantada GL, Luna-Fineman S, Qaddoumi I et al. Retinoblastoma in developing countries. In: Retinoblastoma. EE.UU.: Springer;2010:133-41.
9. Rodríguez-Galindo C. Retinoblastoma: one world, one vision. Pediatrics. 2008;122(3):763-70.
10. Melamud A, Palekar R, Singh A. Retinoblastoma. Am Fam Phys. 2006;73(6):1039-44.
11. Canturk S, Qaddoumi I, Khetan V, Ma Z, Furmanchuk A, Antoneli CBG, et al. Survival of retinoblastoma in less-developed countries impact of socioeconomic and health-related indicators. Br J Ophthalmol. 2010;94:1432-36.
12. Johns Hopkins University. Online Mendelian Inheritance in Man (OMIM). Baltimore: John Hopkins University; 2014 [citado 14 de diciembre de 2017]. Disponible en: http://omim.org/entry/180200
13. Shields JA, Shields CL, Ehya H, Eagle RC Jr, De Potter P. Fine‑needle aspiration biopsy of suspected intraocular tumors. The 1992 Urwick Lecture. Ophthalmology. 1993;100:1677-84.
14. Singh H, Jain P, Gupta M, Bajaj A. Retinoblastoma: A curse to childhood. Ind J Clin Pract. 2013;24(2):34-9.
15. Singh G, Daniels AB. Disparities in retinoblastoma presentation, treatment, and outcomes in developed and less-developed countries. Semin Ophthalmol. 2016;31(4):3106.
16. Gallie BL, Squire JA, Goddard A, Dunn JM, Canton M, Hinton D, et al. Mechanism of oncogenesis in retinoblastoma. Lab Invest. 1990;62:394-408.
17. Gallie BL, Ellsworth RM, Abramson DH, Phillips RA. Retinoma: Spontaneous regression of retinoblastoma or benign manifestation of the mutation? Br J Cancer. 1982;45:513-21.
18. Gallie BL, Phillips RA, Ellsworth RM, Abramson DH. Significance of retinoma and phthisis bulbi for retinoblastoma. Ophthalmology. 1982;89:1393‑9.
19. Mills MD, Syed N. Retinoblastoma in an eye with congenital uveal coloboma. J Am Assoc Pediatr Ophthalmol Strab. 1998;2:303-4.
20. Manzitti J, Mansilla MC. Retinoblastoma. Arch Argent Pediatr. 2010;108(3):255-7.
21. Ah Chu M. Tumores Infantiles: dirigido al médico de atención primaria. Panamá: Editora Offsetcolor; 2001.
22. Trapatsas C, Ah Chu M. Tumores orbitarios en niños. Panamá: Rev Méd Cient. 2006;15(2):103-7.
23. Zhang X, Zheng L, Gao F, Dong F. Adult Onset Retinoblastoma: A Case Report. Chin Med J. 2015;128:133-4.
Recibido: 21 de febrero de 2018.
Aprobado: 1ro. de marzo de 2018.
Liudmira González Rodríguez. Hospital Pediátrico "Juan Manuel Márquez". La Habana, Cuba. Correo electrónico: liudmilagr@infomed.sld.cu

