










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El glaucoma congénito primario (GCP) o del desarrollo, de comienzo temprano, es denominado en dependencia de la edad de comienzo. Se le llama congénito si está presente al nacimiento o en los primeros 3 meses de vida; infantil de los 3 meses hasta los 3-4 años; y juvenil después y hasta los 35 o más años de vida. Es producto del aumento de la presión intraocular (PIO), condicionado por anormalidades en el desarrollo del ángulo de la cámara anterior (CA) intraútero, con manifestaciones oculares en el recién nacido y en el niño, que originan alteraciones morfológicas del globo ocular y afectación del nervio óptico, lo cual puede provocar baja visión o ceguera en etapas tan tempranas como el nacimiento o en los primeros meses o años de vida.1,2,3,4,5,6,7
Estos glaucomas se manifiestan tempranamente porque las estructuras del ángulo no han adoptado una disposición definitiva al nacimiento, por lo que reciben el nombre de glaucomas del desarrollo de comienzo temprano (GDCT), aunque existen formas precoces y otras más tardías. Si las alteraciones morfológicas angulares son leves, su aparición puede retrasarse hasta la adolescencia, denominado glaucoma juvenil, o aparecer incluso más tardíamente (glaucoma juvenil tardío).5,6
Consideraciones embriológicas
Las estructuras del segmento anterior del globo ocular tienen diferente origen embriológico. Para citar solamente las que intervienen en la formación del ángulo de la CA y la salida del humor acuoso del ojo, el epitelio corneal y el cristalino derivan del ectodermo superficial; del neuroectodermo lo hacen los músculos esfínter y dilatador del iris. De su epitelio pigmentario, la zónula, el cuerpo ciliar y el epitelio ciliar no pigmentado y de las células de la cresta neural craneal provienen el estroma y el endotelio corneal, la esclera (excepto su porción temporal), la malla trabecular, el tejido conectivo del iris, el canal de Schlemm, el ángulo camerular, el estroma de coroides y el músculo del cuerpo ciliar. Esto explica la gran prevalencia de glaucoma asociada al amplio espectro de anomalías del desarrollo, también conocidas como disgenesias del segmento anterior o mesenquimatosas, las que fueron agrupadas antiguamente como síndrome de clivaje del segmento anterior.1,7,8,9,10,11,12,13
Clasificación
Epidemiología y herencia
La herencia se plantea como autosómica recesiva, con penetrancia incompleta, poligénica o multifactorial, con variadas alteraciones genéticas. Se han identificado 4 locus de cromosomas afectados: GLC3A en 2p21; GLC3B en lp36; GLC3C en 14q24.3; y GLC3D en 14q24.3. Tienen alta incidencia de GCP las poblaciones con alto índice de consanguinidad, especialmente las que tienen el gen CYP1B1.
El glaucoma juvenil de ángulo abierto tiene una herencia autosómica dominante y ha sido relacionado con el GLC1A myocilin gene (MYOC), responsable de algunos glaucomas de ángulo abierto del adulto. Aunque hemos señalado cómo se agrupan habitualmente todos los glaucomas en el niño, solamente nos referiremos al glaucoma congénito primario.
GLAUCOMA CONGÉNITO PRIMARIO
El glaucoma congénito primario es originado por una trabéculo-disgenesia aislada (sin otras manifestaciones oculares o sistémicas). El diagnóstico se realiza al nacimiento. Solamente en el 25 % de los pacientes afectados y en más del 80 % se diagnostica en el primer año de vida. Se presenta en el sexo masculino en el 65 % de las ocasiones. Es bilateral en el 70-75 %, aunque puede presentarse de forma asimétrica y en un ojo más tardíamente que en el otro. Su incidencia es variable en las diversas áreas del mundo. En Occidente se describe 1: 10 000 nacidos vivos y en medio Oriente: 1: 2 500.5,6 El glaucoma neonatal y el de comienzo o reconocimiento tardío se asocian a peor pronóstico.
Cuadro clínico. Síntomas y signos
El niño pequeño no puede expresar sus molestias, por lo que es importante tener en cuenta lo referido por la madre u otro familiar, como la presencia de "ojos opacos", rechazo a salir a la luz intensa, lagrimeo, entre otros. En estos casos, el examen oftalmológico minucioso cobra más valor y nos conduce a realizar la exploración, que debe ser bajo sedación o anestesia general, para lo cual también requeriremos de una valoración sistémica por Pediatría.11,12,13,14,15,16,17,18,19
Signos inespecíficos: La triada característica de alarma del segmento anterior (lagrimeo, fotofobia y blefarospasmo) son producto del edema corneal epitelial por aumento de la PIO.11
Signos específicos: Alteraciones de la transparencia corneal, aumento de tamaño de la córnea, del limbo corneal y del globo ocular, aumento de profundidad de la cámara anterior y miopización entre otros, ocasionados por la gran elasticidad de los tejidos oculares en estas etapas de la vida.6,19
Conducta a seguir
La conducta a seguir es la exploración bajo anestesia y si se confirma el diagnóstico se procede a la cirugía.
Exploración
La exploración bajo anestesia general tiene una baja tasa de riesgo, por el desarrollo de nuevos fármacos y máquinas de anestesia. Los agentes anestésicos son hipotensores locales en su mayoría,2,6 excepto la ketamina y la acetilcolina, que elevan la PIO. No olvidar que cuando a estos pacientes se les realiza laringoscopia e intubación, estas maniobras pueden elevar la PIO.2
Corneometría: Es necesario realizar la medición del diámetro corneal horizontal (corneometría), que es aproximadamente 0,5 mm mayor que el vertical,1 con cifras consideradas como normales de 9,5 a 10,5 mm al nacimiento. Cifras por encima de 11,5 mm son sugerentes de glaucoma. Al año de edad, el valor referencial es de 10 a 11,5 mm, con cifras por encima de 12,5 mm. Hay que estar alertas y sospecharlo siempre que tenga más de 13,0 mm de diámetro corneal.18
Biomicroscopia: En la que apreciaremos los signos oftalmológicos específicos, además del aumento del diámetro corneal y del limbo corneoescleral; la transparencia de la córnea, si existe edema u opacidades corneales (estrías de Haab en casos avanzados); profundidad de la CA (puede alcanzar hasta 7,3 mm);1 iridodonesis y subluxación del lente, entre otras.19
Tonometría: Debe realizarse con tonómetro de aplanación manual, y las cifras de la PIO consideradas normales estarán oscilando hasta 15 mmHg, mientras que una presión entre 15 a 20 mmHg se considerará como dudosa y se confirma el diagnóstico con cifras tensionales por encima de 20 mmHg, aunque en la actualidad existen autores como Allingham y otros20 que discrepan de estos valores, por lo que los resultados de esta exploración siempre estarán muy relacionados con la sintomatología y los signos oftalmológicos que presenta el paciente.20 El espesor corneal central es mayor en el niño con hipertensión ocular que en el adulto con glaucoma, por lo que el valor de la PIO puede estar falsamente elevado.14 Los tonómetros más usados en niños y jóvenes para realizar la exploración son: Tono-Pen (Reichert Ophthalmic Instruments, Depew, NY); Icare (Icare Finland Oy, Helsinki, Finland) y Perkins (Haag-Streit EE.UU., Mason, OH).
Gonioscopia: Las alteraciones del ángulo se agrupan fundamentalmente en 3 grupos (cuadro), según la anomalía del tejido (descritas por Luntz en el año 1979) y acorde con la localización anatómica de los tejidos (descritas por Hoskins en el año 1983), y juegan un papel primordial en el diagnóstico y pronóstico de la enfermedad.1
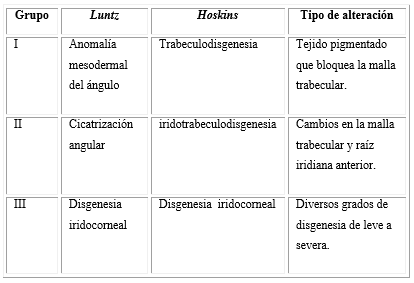
Con la gonioscopia, si la transparencia de los medios lo permite, podremos observar las características y las posibles alteraciones del ángulo, como membrana de aspecto blanquecino, de Barkan (con aspecto de celofán); presencia de restos mesodérmicos; engrosamiento de la malla trabecular; inserción anterior del iris; ausencia de receso angular o hasta un ángulo de aspecto normal.2,4,6
En las alteraciones anatomo-histológicas puede presentarse hipoplasia del espolón escleral y del iris, inserción anterior iridiana y de fibras longitudinales del músculo ciliar en la malla uveal, tejido reticular pigmentado, capa celular sobre malla trabecular, alteración en la densidad de espacios reticulares, colapso o ausencia del canal de Schlemm, algunas de ellas observables a la gonioscopia.10
Oftalmoscopia: Si la excavación papilar es mayor de 0,3 en un lactante, es muy sugestiva de glaucoma congénito. Podemos encontrar palidez y excavación papilar aumentada, que varía acorde con los cambios de la PIO, por el aumento de la elasticidad de los tejidos, y puede revertirse con un control tensional precoz y adecuado.1,6
Biometría: Considerado como normal, el eje axial tiene valores en el recién nacido de 17 mm; 19,10 mm al año; 19,60 mm a los 2 años; 20,50 mm a los 6 años y 23,90 mm a los 15 años, con una variación de ± 0,25.2,18,19 La biometría tiene valor en las primeras etapas de la vida, ya que después de los 4 a 5 años de edad el ojo del niño no sigue creciendo como consecuencia del aumento de la PIO por el incremento de la resistencia corneoescleral.2
Biomicroscopia ultrasónica (BMU): Por medio de ella se pueden obtener imágenes de alta resolución de la córnea, la cámara anterior, el ángulo, el iris, el cuerpo ciliar y la cámara posterior del globo ocular, las cuales son de mucha ayuda para decidir la cirugía en presencia de opacidades corneales densas, en las que no se puede apreciar el segmento anterior ni los detalles a la gonioscopia.3
Tomografia confocal y estudio del campo visual: Tendrán valor cuando el niño sea lo suficientemente mayor para cooperar al examen.
Diagnóstico diferencial
Se realiza con las entidades que provocan aumento de las dimensiones corneales y aquellas que ocasionan lagrimeo y fotofobia:
Megalocórnea congénita: aislada o asociada a otras anomalías del segmento anterior. En su manifestación aislada, consiste en un alargamiento corneal no progresivo, con un diámetro corneal mayor de 12 mm en el recién nacido o mayor de 13 mm a cualquier edad, sin aumento de la presión intraocular, ni rupturas de la membrana de Descemet.18 bLa megalocórnea congénita puede asociarse a otras anomalías oculares que incluyen pigmentación a nivel de la malla trabecular, glaucoma de comienzo tardío, hipoplasia del estroma iridiano, catarata, ectopia lentis, entre otros, y entre las asociaciones sistémicas están los síndromes de Alport, Marfán, Down, retardo mental y megalocórnea, mucolipidosis tipo II y craneosinostosis. La megalocórnea ha sido categorizada en cuatro tipos:11
Tipo 1: Síndrome de Neuhauser (megalocórnea, hipoplasia de iris y retardo mental).
Tipo 2: Megalocórnea asociada a escoliosis y retardo del crecimiento.
Tipo 3: Megalocórnea asociada a hipotonía severa y microcefalia.
Tipo 4: Megalocórnea asociada con megaloencéfalo y obesidad.
Generalmente pueden existir otros familiares cercanos (madre, padre), con semejantes características oculares. Se reporta asociación entre las regiones DXS87 y DXS94 en los cromosomas Xq21.3-q22 y la megalocórnea.4) La miopía es el defecto refractivo más común asociado a esta condición, así como el astigmatismo a favor de la regla. Es necesaria la observación y el seguimiento periódico para detectar complicaciones y la presencia de un glaucoma de comienzo tardío. También es necesario diferenciarlo con:
Tratamiento
La reducción de la presión intraocular es el objetivo del tratamiento1 en este y en cualquier otro glaucoma, para evitar el deterioro del nervio óptico y conservar la visión, ya que hasta el momento la PIO es el único factor de riesgo sobre el que podemos actuar. 2,21,22
El tratamiento médico es transitorio y necesario para reducir la presión ocular y el edema corneal previo a la cirugía. También es utilizado como tratamiento de soporte en glaucomas descompensados después de la cirugía intraocular y en los refractarios a tratamiento convencional.6
Si la PIO es menor de 15 mmHg se descarta el diagnóstico; si es menor de 15 mmHg pero existen otros signos o la PIO oscila entre 15 y 20 mmHg sin signos, debemos repetir la exploración. Si la PIO está entre 15 y 20 mmHg y se acompaña de signos, el diagnóstico estará confirmado y se realizará cirugía, al igual que con presiones oculares por encima de 20 mmHg.2,22
La cirugía es el tratamiento de elección, definitivo e inicial, principalmente a nivel del ángulo de la CA, con variantes como: goniotomía, trabeculotomía, trabeculo-trabeculectomía con o sin antimetabolitos, canaloplastia con prolene, trabeculotomía transluminal asistida por goniotomía, viscocanalostomía, trabeculotomía con microcatéter iluminado, dispositivo de drenaje con o sin antimetabolitos y en última instancia los procedimientos ciclodestructivos.1,4,8,10,13,14,21,22 En el ángulo cicatrizal y en las disgenesias iridocorneales, el procedimiento combinado de trabéculo-trabeculectomía logra mejores resultados que la trabeculotomía y la goniotomía.1,19,23)
Pronóstico
El pronóstico es pobre para controlar la PIO y garantizar la visión si el glaucoma está presente al nacimiento, el tamaño del diámetro corneal está por encima de 14 mm al momento de realizar el diagnóstico y si el tratamiento quirúrgico fue demorado por alguna causa.2,6
Rehabilitación
La rehabilitación visual de estos pacientes es de gran importancia después de la cirugía una vez controlada la hipertensión ocular. Es necesario cuidar del desarrollo visual del niño, que puede alterarse o interrumpirse por cambios en la estructura anatómica del ojo y/o lesión orgánica residual asociada o producto de este.
Para lograr una rehabilitación adecuada y un mejor pronóstico visual, debemos tener en cuenta los múltiples factores que pueden intervenir en la disminución visual de estos niños, como son las cicatrices y opacidades corneales, el daño del nervio óptico, la miopía en pacientes que presentan glaucoma en los tres primeros años de vida como consecuencia del aumento secundario de la longitud axial por PIO elevada, el astigmatismo producto de las estrías de Habb o resultante del tratamiento quirúrgico, la asociación de los trastornos refractivos a anisometropía y la ambliopía estrábica, más frecuentes en las formas unilaterales.2,18,19,24,25
La ambliopía puede presentarse más comúnmente en la forma monocular,2 pero también bilateral, precozmente (antes de los 3 - 5 años), o más tardía (posterior a los 5 años de edad) y puede concomitar con anisometropías, astigmatismos mayores de 3 dioptrías y alteraciones motoras.2
CONSIDERACIONES FINALES BASADAS EN LA EXPERIENCIA
Con la goniotomía o la trabeculotomía obtuvimos poca efectividad en nuestro medio, lo cual relacionamos con el hecho de que en las décadas del 80 y el 90 el Instituto Cubano de Oftalmología “Ramón Pando Ferrer” fue el Centro receptor de prácticamente todos los glaucomas infantiles del país. Recibió a pacientes con alteraciones oculares marcadas, producto de glaucoma congénito primario, muchos de los cuales no acudían precozmente por la lejanía geográfica y las condiciones precarias del transporte, lo que nos llevó a elegir la trabéculo-trabeculectomía como procedimiento quirúrgico inicial en todos los pacientes, sin tomar en cuenta la transparencia corneal ni la asociación de alteraciones angulares como la cicatrización, la no permeabilización, la atrofia o la ausencia del canal de Schlemm, ya que en esos momentos no contábamos con técnicas de imagen como la BMU.
Por igual razón, si estaba indicada la cirugía y no existían complicaciones transoperatorias ni contraindicaciones anestésicas, operábamos ambos ojos en el mismo acto quirúrgico23) para evitar la demora en el regreso de los pacientes por vivir en otras provincias del país, así como por episodios respiratorios agudos ocasionados por la estancia en la unidad quirúrgica y por la anestesia, factores que retrasaban mucho la intervención del segundo ojo, lo que ensombrecía el pronóstico visual.
Las complicaciones transoperatorias con esta variante quirúrgica fueron mínimas, del tipo de hipemas o aplanamiento de la CA, que resolvían en pocos días con tratamiento médico.
Consideramos que esta técnica quirúrgica es de gran efectividad si el canal de Schlemm25 se encuentra permeable, pues permite la salida del humor acuoso por un doble mecanismo de acción: elimina el obstáculo a nivel del trabéculo y el canal de Schlemm, y corta el tejido anómalo del ángulo (filtración interna), además de permitir una vía de filtración externa al espacio subconjuntival18) por medio de la trabeculectomía, en caso de no estar permeable el Schlemm.25
Recomendaciones
Realizar el diagnóstico lo más tempranamente posible, para lo que es necesaria la instrucción de pediatras y oftalmólogos generales con vistas a lograr la remisión adecuada y precoz del niño a un centro especializado para el tratamiento quirúrgico, con lo cual mejora su pronóstico visual.
Realizar la cirugía de ambos ojos si está indicada y no existen complicaciones transquirúrgicas ni anestésicas.
Mantener estrecho control refractivo y realizar la corrección de anisometropías para prevenir ambliopías.
Concientizar a la familia y al paciente de la necesidad de un seguimiento de por vida.

