Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Archivo Médico de Camagüey
versión On-line ISSN 1025-0255
AMC vol.15 no.5 Camagüey sep.-oct. 2011
ARTÍCULOS ORIGINALES
Aspectos médicos, genéticos y psicosociales del síndrome Usher
Medical, genetic, and psychosocial aspects of Usher´s syndrome
Dra. Elisa Dyce GordonI; Dra. Yolanda Mapolón ArcendorII; Dr C. Jorge Santana ÁlvarezIII
II Especialista de II Grado en Oftalmología. Centro Provincial de Retinosis Pigmentaria. Camagüey, Cuba.
III Doctor en Ciencias. Especialista de II Grado en Otorrinolaringología. Investigador Agregado. Hospital Militar Octavio de la Concepción y la Pedraja. Camagüey, Cuba. jorsan@finlay.cmw.sld.cu
RESUMEN
Fundamento: el síndrome Usher es una enfermedad genética, que se caracteriza por hipoacusia neurosensorial progresiva bilateral congénita, pérdida de visión debida a la retinosis pigmentaria y en ocasiones presenta también trastornos vestibulares. Objetivo: describir los principales aspectos médicos, genéticos y psicosociales presentes en los pacientes con síndrome Usher. Método: se realizó un estudio descriptivo transversal en 14 pacientes con diagnóstico de síndrome Usher atendidos en el Centro de Retinosis Pigmentaria de Camagüey, desde 1991 hasta 2008. Resultados: los 14 pacientes provenían de familias diferentes y seis de ellos tenían antecedentes patológicos familiares. Se encontró consanguinidad entre los padres de las personas afectadas en el 35,72 % de las familias, el principal grado de parentesco fue el de primos hermanos. El síndrome Usher tipo II fue la forma más frecuente. La mayoría de los pacientes eran procedentes de zonas urbanas; los que portaban el síndrome levemente aspiraban a alcanzar un mayor nivel de escolaridad, así como aquellos procedentes de las áreas urbanas. Tenían ocupación laboral nueve; algunos con trabajos riesgosos. De los nueve, cuatro no trabajaban y sólo dos eran del sexo femenino. Conclusiones: las manifestaciones clínicas del síndrome Usher provocan una doble limitación (visual y auditiva), que son responsables de los trastornos psicológicos y sociales que se asocian al mismo. Las personas afectadas necesitan de una atención especial que contribuya a mejorar la calidad de sus vidas.DeSC: SINDROMES DE USHER/genética; TRASTORNOS SORDOCEGUERA; PÉRDIDA AUDITIVA SENSORINEURAL; ESTUDIOS TRANSVERSALES.
ABSTRACT
Background: Usher´s syndrome is a genetic disease that is characterized by congenital bilateral progressive neurosensory hypoacusis, loss of vision due to retinitis pigmentosa and occasionally also presents vestibular disorders. Objective: to describe the main medical, genetic, and psychosocial aspects present in patients with Usher´s syndrome. Method: a cross-sectional descriptive study was conducted in 14 patients with Usher´s syndrome diagnosis treated at the pigmentary retinosis Center in Camagüey, from 1991 to 2008. Results: fourteen patients came from different families and six of them had family pathological history. Found consanguinity among parents of persons affected by 35,72 % of families, cousins was the main degree of kinship. Usher´s syndrome type II was the most common form. The majority of patients were from urban areas; which carried the syndrome slightly aspired to achieve a higher level of schooling, as well as those from urban areas. Nine of them had jobs, some risky works. Four out of nine, did not work and only two were female. Conclusions: clinical manifestations of Usher´s syndrome cause a double constraint (visual and auditory), which are responsible for the psychological and social disorders that are associated to it. Affected persons need a special care to help improve the quality of their lives. DeSC: USHER SYNDROME/genetics; DEAF-BLIND DISORDERS; HEARING LOSS, SENSORINEURAL; CROSS-SECTIONAL STUDIES. INTRODUCCIÓN El síndrome Usher (SU) es una enfermedad genética, que se caracteriza por hipoacusia neurosensorial progresiva bilateral congénita, pérdida de visión debida a la retinosis pigmentaria (RP) y en ocasiones presenta también trastornos vestibulares.1-4 Bell, referido por Kimberling, et al, 1 en 1933 fue uno de los primeros en identificar la heterogeneidad clínica que caracteriza a este síndrome, debida a su heterogeneidad genética. El carácter hereditario del SU es conocido desde 1914.5 Epidemiológicamente se conoce como la primera causa de sordo-ceguera en el humano.6 Del 3 al 6 % de las personas sordas y el 5 % de los niños sordos es debido a este síndrome, el cual es la causa más común e identificable de sordera hereditaria entre niños profundamente sordos.1, 2 Desde que se inició el Programa Nacional de Retinosis Pigmentaria en Cuba en 1990,7 el estudio de este síndrome aumenta no solo por los otorrinolaringólogos, sino por oftalmólogos y genetistas, por ser este síndrome el más asociado a la RP.8, 9 Su prevalencia en Cuba es de 4.4 por cada 100 000 habitantes y en Camagüey es aproximadamente de 2.78 por cada 100 000 habitantes.8 Las personas afectadas están doblemente limitadas, por el déficit auditivo y por el visual, y en ocasiones por las alteraciones del equilibrio. Debido al trastorno auditivo pueden presentar alteraciones en el desarrollo del lenguaje; y si no son rehabilitados temprana y adecuadamente pueden llegar hasta el mutismo. También se ha observado en estos pacientes, carencia intelectual, aislamiento social, alteraciones psicológicas, psiquiátricas y neurológicas que producen sufrimiento, frustración y miedo a la persona afectada y a su familia. Existen diversidades de reportes clínicos, genéticos, moleculares acerca de este síndrome, pero no integran los aspectos clínicos y genéticos con los psicológicos y sociales, que son muy importantes. En Cuba hay pocos reportes del síndrome. Debido a la importancia del mismo surgió la motivación para realizar este estudio, con el objetivo de describir algunos aspectos médico-genético, psicológicos y sociales relacionados al SU que contribuyan a aumentar el nivel de conocimiento acerca del mismo, a orientar el trabajo de los equipos multidisciplinarios y a la elaboración de proyectos de atención médico-social específicos para los mismos. MÉTODOS El universo estuvo constituido por 21 pacientes. La muestra no probabilística la conformaron 14 pacientes con el diagnóstico del síndrome e historias clínicas completas. Los siete pacientes restantes fueron excluidos del estudio por datos insuficientes en las historias clínicas. Todos los pacientes fueron valorados por el equipo multidisciplinario. El diagnóstico y clasificación clínica del síndrome fue realizado por un especialista en otorrinolaringología por medio de estudio clínico, audiológico y vestibular. Fueron revisadas las historias clínicas oftalmológicas y genéticas. Esta última incluye el interrogatorio, examen físico general y confección del árbol genealógico. Se recogieron datos biológicos y sociales, tales como nombres y apellidos, edad, sexo, dirección, área de salud, escolaridad, ocupación, procedencia urbana o rural, así como la clasificación clínica, aspectos psicológicos, tipo de herencia, consanguinidad y familiares afectados; los cuales fueron vertidos en un modelo confeccionado para ese fin. Los datos fueron procesados por técnicas de estadística descriptiva, hallándose distribuciones de frecuencias expresadas en por cientos. Fueron presentadas en forma de tablas. RESULTADOS Se estudiaron 14 personas provenientes de 14 familias. De ellos, seis tenían antecedentes patológicos familiares (42,85 %). Se encontró consanguinidad entre los padres de las personas afectadas en el 35,72 % de las familias (N=5), donde el principal grado de parentesco fue el de primos hermanos. (Tabla 1)
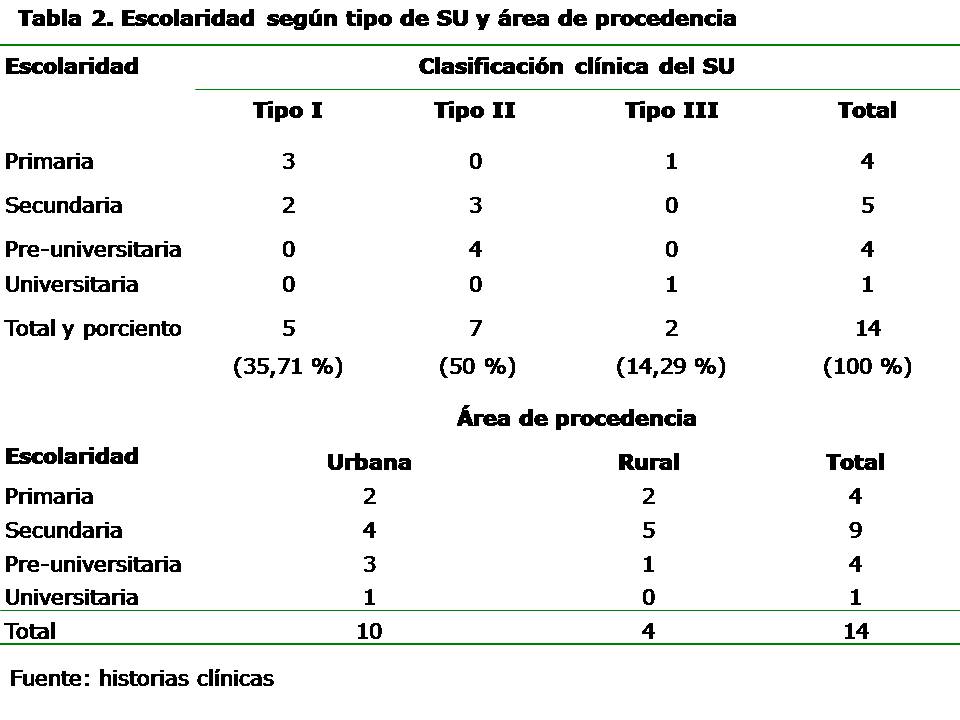
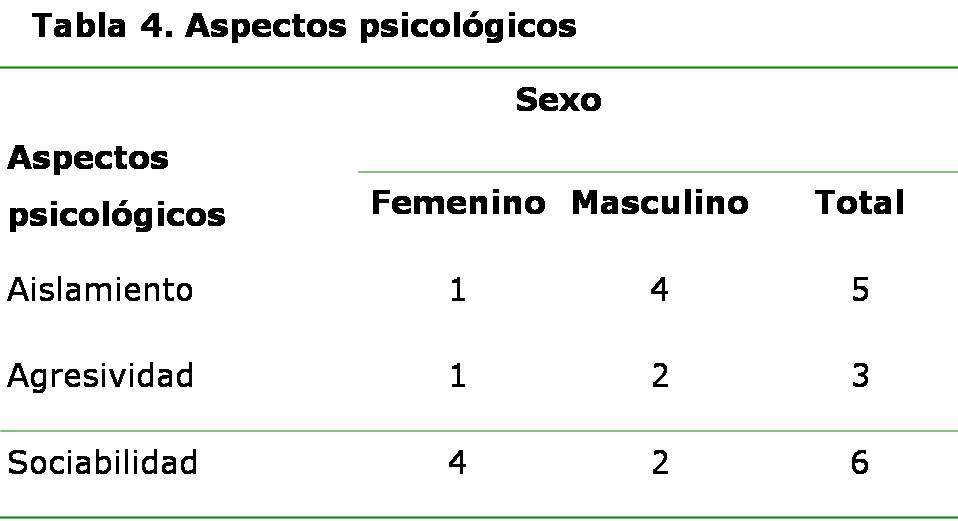
Se diagnosticaron las tres formas del SU descritas en la literatura. El SU tipo II fue la forma más frecuentemente encontrada, seguido por el tipo I. Se encontraron personas en el nivel primario, secundario y pre-universitario. Con respecto a la relación entre escolaridad y tipo de SU, se encontró que el nivel de escolaridad es mayor en los tipos II y III. Sólo se encontró a un paciente con nivel de escolaridad universitario, el cual presentó la forma leve del SU (tipo III). Las personas con el SU tipo I no pasaron de la enseñanza secundaria y los del tipo II estaban entre secundaria y preuniversitario. La mayoría de los pacientes eran procedentes de zonas urbanas (10/14) (71,42 %), sin embargo, cabe destacar que la mayoría de los procedentes de zonas rurales alcanzaron llegar a la enseñanza secundaria aunque no la terminaron. Solo uno alcanzó la enseñanza preuniversitaria. (Tabla 2) Tenían ocupación nueve pacientes para un 64,28 %, la mayoría con trabajos que ponen en riesgo la salud e incluso la vida, como es el caso de un albañil, un obrero agrícola y un oficial de seguridad y protección. De los nueve ocupados, sólo dos son del sexo femenino y del total de mujeres (N=6), cuatro no trabajan. Todas están en edad laboral, pero con severa hipoacusia y RP en estadios III y IV. (Tabla 3) Se detectaron aspectos psicológicos tales como la tendencia al aislamiento en el sexo masculino y a la sociabilidad en el sexo femenino. (Tabla 4) DISCUSIÓN Este es un tipo de herencia horizontal donde las personas afectadas son de la misma generación, fundamentalmente hermanos, y en muchas familias se observa sólo una persona afectada lo cual concuerda con nuestra investigación. La consanguinidad o parentesco entre los padres es un importante factor genético y social que aumenta el riesgo de presentación de las enfermedades genéticas; en el primer caso por aumentar la probabilidad de formar genotipos homocigóticos recesivos y en el segundo caso por el aumento en la aparición de las enfermedades genéticas. La heterogeneidad genética del síndrome, con la participación de diferentes genes y mutaciones en la producción del fenotipo, es responsable de las diferentes formas clínicas existentes. 11-14 Inicialmente, se consideraron dos formas clínicas de la enfermedad, no obstante, en la actualidad se reconocen tres formas clínicas con al menos nueve genes identificados. 15-16 El SU tipo I caracterizado por sordera sensorineural de profunda a severa, congénita, bilateral y simétrica, no progresiva; con inicio de la RP entre los ocho y los 15 años de edad así como respuesta vestibular anormal. El SU tipo II presenta sordera sensorineural leve, moderada leve, moderada o severa, congénita bilateral, simétrica, no progresiva. El inicio de la RP es después de los 15 años de edad, y la respuesta vestibular es normal o anormal. 1 En el tipo III hay un grado de afectación visual por la RP y disfunción vestibular variables, combinada con pérdida auditiva progresiva. 6 Estas formas clínicas difieren en la edad de aparición de la RP, la evolución y severidad clínica. Esta clasificación clínica del SU es de gran valor para la integración social de estas personas, ya que en el tipo I las personas tienden a quedar completamente sordas, a veces mudas, ciegas o débiles visuales a muy temprana edad. Esta discapacidad auditiva que aparece fundamentalmente durante las primeros dos o tres años de vida, afecta notablemente el desarrollo del lenguaje, el cual se adquiere de forma espontánea al escuchar el habla de otras personas y la de uno mismo. A veces estas personas quedan mudas así como ciegas o débiles visuales a muy temprana edad, mientras que en el tipo II las personas son hipoacúsicas y las manifestaciones oftalmológicas aparecen después de los 12 años; y III el grado de hipoacusia y debilidad visual aparecen más tardíamente. En ambos tipos (II y III) se afecta menos el lenguaje y ofrece mayores posibilidades de integración social. Independientemente de que en Cuba todas las personas tienen las mismas oportunidades sociales de educación, existen diferencias entre las personas con desiguales tipos de SU debido a las características clínicas presentes en cada uno de ellos. Menos posibilidades biológicas en el tipo I y mayores en los tipos II y III, ya que el grado de escolaridad que alcanzan estas personas depende en gran parte de la clasificación clínica. Hay escuelas tanto en zonas urbanas como rurales, pero las personas afectadas por esta enfermedad tienen ciertas dificultades inherentes a su problema de salud, fundamentalmente debidas a la nictalopía que les impiden desempeñarse al igual que sus compañeros, el hecho de vivir en áreas rurales o en zonas con menor cantidad de luz. El mayor número de hombres con ocupación laboral al igual que los aspectos psicológicos descritos pudieran estar relacionados con los roles de género, ya que tradicionalmente la sociedad le ha asignado a la mujer el papel de protectora de la familia, y prefieren quedarse en casa atendiendo a los hijos, esposos y a ellas mismas, más aún con una enfermedad discapacitante. Las mujeres también resultan más dóciles y sociables, mientras los hombres ante la frustración dada por la enfermedad tienden a aislarse y en ocasiones refieren ocupar su tiempo libre ingiriendo bebidas alcohólicas, que puede aumentar su grado de discapacidad visual y el riesgo de accidentes. El déficit de audición y visión disminuyen considerablemente la calidad de vida que repercute en el desarrollo del individuo, ya que afecta la comunicación, el intelecto, y la integración a la sociedad. La disminución de estos estímulos sensoriales también provoca alteraciones de la personalidad, estados de angustia y trastornos emocionales. Estas personas tienen serias dificultades en sus relaciones sociales y afectivas, se sienten diferentes y se aíslan. Se han reportado en otros estudios trastornos psicóticos en el SU, así como anomalías morfológicas del sistema nervioso central. 17-19 CONCLUSIONES Múltiples son los aspectos médico-genéticos, psicológicos y sociales asociados al SU, entre ellos, la herencia, el tipo de SU, la escolaridad, la ocupación, características psicológicas, entre otras. Los pacientes portadores de esta enfermedad se ven limitados escolarmente, pocos llegan a la educación superior, tienen una inadecuada ubicación laboral, carecen de ocupación de su tiempo libre y sufren por trastornos psicológicos. REFERENCIAS BIBLIOGRÁFICAS 1. Kimberling WJ, Möller C. Clinical and molecular genetics of Usher syndrome. J Am Acad Audiol. 1995; 6:63-72. 2. Young NM, Mets MB, Haim TC. Early diagnosis of Usher syndrome in infants and children. Am J Oto. 1996; 17:30-4. 3. Jaijo T, Aller E, Beneyto M, Najera C, Millan JM. Molecular genetic study of Usher syndrome in Spain. Acta Otorrinolaringol Esp. 2005 Feb; 56(2):55-8. 4. Bernal S, Meda C, Solans T, Ayuso C, Garcia-Sandoval B, Valverde D, et al. Clinical and genetic studies in Spanish patients with Usher syndrome type II: description of new mutations and evidence for a lack of genotype--phenotype correlation. Clin Genet. 2005 Sep; 68(3):204-14. 5. Reiners J, Nagel-Wolfrum K, Jurgens K, Marker T, Wolfrum U. Molecular basis of human Usher syndrome: deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp Eye Res. 2006 Jul; 83(1):97-119. 6. Tamayo ML. La sordo-ceguera: Un reto actual. En: Tamayo ML. Manual de genética de la retinitis pigmentosa y el síndrome de Usher. Colombia: INCI; 1996.p.5-6. 7. Colectivo de autores. Retinosis pigmentaria y sordera en Cuba. Av Med Cuba. 2003; Año X(34):4-7. 8. Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006 Oct 11; 1:40. 9. Dyce B, Mejías J, Copello M, Hernández R, Horrach I. Aspectos genéticos y clínicos del síndrome de usher. Rev Cubana Oftalmol. 2000; 13(2):79-83. 10. Oti M, Brunner H. The modular nature of genetic diseases. Clin Genet. 2007 Jan; 71(1):1-11. 11. Tamayo ML, Lopez G, Gelvez N, Medina D, Kimberling WJ, Rodríguez V, et al. Genetic counseling in Usher syndrome: linkage and mutational analysis of 10 Colombian families. Genet Couns. 2008; 19(1):15-27. 12. Najera C, Beneyto M, Millán JM. Usher syndrome: an example of genetic heterogeneity. Med Clin (Barc). 2005 Oct 1; 125(11):423-7. 13. Seeliger MW, Fischer MD, Pfister M. Usher syndrome: clinical features, diagnostic options, and therapeutic prospects. Ophthalmologe. 2009 Jun; 6(6):505-11. 14. Bolz HJ. Genetics of Usher syndrome. Ophthalmologe. 2009 Jun; 106(6):496-504. 15. Yan D, Liu XZ. Genetics and pathological mechanisms of Usher syndrome. J Hum Genet. 2010 Apr 9; 3(5):23-35. 16. Wu CY, Chiu CC. Usher syndrome with psychotic symptoms: two cases in the same family. Psychiatry Clin Neurosci. 2006 Oct; 60(5):626-8. 17. Rijavec N, Grubic VN. Usher syndrome and psychiatric symptoms: a challenge in psychiatric management. Psychiatr Danub. 2009 Mar; 21(1):68-71. 18. Viala A, Nicot T, Levy F, Vacheron MN. A case of Usher's syndrome associated with psychotic symptoms: diagnosis and follow-up in a psychiatric unit. Encephale. 2009 Jun; 35(3):286-91. 19. Demir HD, Deniz FE, Yardim H. A rare brain developmental anomaly in a patient with Usher's syndrome. Int Ophthalmol. 2010 Feb; 30(1):85-8. Recibido: 20 de septiembre de 2010 Aprobado: 29 de junio de 2011 Dra. Elisa Dyce Gordon. Email: dgelisa@finlay.cmw.sld.cu