










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Archivo Médico de Camagüey
versión On-line ISSN 1025-0255
AMC vol.17 no.3 Camagüey mayo-jun. 2013
CASOS CLÍNICOS
Síndrome poliglandular autoinmune tipo II: presentación de un caso
Polyglandular autoimmune syndrome type II: a case report
Dr. Rafael Pila Pérez; Dr. Rafael Pila Peláez; Dr. Víctor A. Holguín Prieto; Dr. Etelívar Torres Vargas; Dra. Mariela Rodríguez Martí
Hospital Universitario Manuel Ascunce Domenech. Camagüey, Cuba.
RESUMEN
Fundamento: la afección poliglandular autoinmune constituye una rareza clínica; de todas las variantes, el síndrome poliglandular autoinmune tipo II (SPGA-II) es el más común, el cual está caracterizado fundamentalmente por la presencia de la enfermedad de Addison autoinmune combinada con tiroiditis autoinmune y diabetes mellitus tipo 1. Este síndrome ocurre fundamentalmente alrededor de la tercera y cuarta décadas de la vida, donde se reporta comúnmente en mujeres con relación a los hombres, con una proporción de 3-4:1.
Objetivo: presentar un caso clínico de un síndrome de Schmidt-Carpenter.
Caso clínico: paciente femenina de 35 años de edad que presenta a los 20 años diabetes mellitus tipo 1, seguida nueve años después de una tiroiditis de Hashimoto, y en los últimos cincos años amenorrea alternando con metrorragias y tres abortos espontáneos; en el ingreso hospitalario se constata alopecia y un complejo sintomático compatible con insuficiencia suprarrenal. Los estudios clínicos y analíticos comprobaron la presencia de un síndrome de Schmidt-Carpenter asociado a hipogonadismo y alopecia.
Conclusiones: este síndrome es una rara enfermedad severa de múltiples glándulas endocrinas causada por trastornos inmunes con destrucción de los tejidos. El diagnóstico es clínico, comprobado por la determinación de los niveles hormonales y las pruebas de inmunidad. Se debe diferenciar de otros procesos inmunes, cromosómicos, hematológicos y digestivos que afectan diferentes glándulas y órganos. La terapéutica empleada fue eficaz. Esta enfermedad es importante para las especialidades clínicas, especialmente la medicina interna, la endocrinología, la inmunología y la genética.
DeCS: POLIENDOCRINOPATÍAS AUTOINMUNES; ENFERMEDAD DE ADDISON; SIGNOS Y SÍNTOMAS; ADULTO; ESTUDIOS DE CASOS.
ABSTRACT
Background: polyglandular autoimmune syndrome type II is a clinical rarity; among all variants this syndrome is the most common. It is mainly characterized by the presence of autoimmune Addison´s disease combined with autoimmune thyroiditis and type I diabetes mellitus. This syndrome appears around the third and fourth decades of life. It is more frequent in women than in men, in a ratio of 3-4:1.
Objective: to present a clinical case of Schmidt-Carpenter syndrome.
Clinical Case: a thirty-five-year-old female patient who presented type I diabetes mellitus at the age of 20 and nine years later a Hashimoto´s thyroiditis. In the last five years the patient presented amenorrhea alternated with metrorrhagia and had three miscarriages. After her hospital admission, it was established alopecia and a symptomatic complex compatible with adrenal insufficiency. Clinical and analytical studies confirmed the presence of Shmidt-Carpenter syndrome associated with hypogonadism and alopecia.
Conclusions: this syndrome is a rare and severe disease that affects multiple endocrine glands caused by immune disorders with destruction of tissues. The diagnosis is clinical, confirmed by the establishment of hormonal levels and immunity tests. This syndrome should be differentiated from other immune, chromosomal, hematological, and digestive processes that affect other glands and organs. The therapeutics employed was effective. This disease is relevant to clinical specialties, particularly for internal medicine, endocrinology, immunology, and genetics.
DeCS: POLYENDOCRINOPATHIES, AUTOIMMUNE; ADDISON DISEASE; SIGNS AND SYMPTOMS; ADULT; CASE STUDIES.
INTRODUCCIÓN
El síndrome poliglandular autoinmune tipo II (SPGA-II) es el más común de los síndromes inmunoendocrinopáticos, el cual es caracterizado por la obligada presencia de la enfermedad de Addison autoinmune en combinación con enfermedad tiroidea autoinmune y/o diabetes mellitus tipo 1.1
El hipogonadismo primario, la miastenia gravis y la enfermedad celíaca son también comúnmente observados en este síndrome.2 La definición de esta enfermedad depende del hecho de que uno de los componentes de estos desórdenes esté presente asociado a otras enfermedades con mayor frecuencia que en la población general. La frecuencia clínica más común de esta combinación es la presencia de la enfermedad de Addison y la tiroiditis de Hashimoto, mientras que la menos frecuente es la sucesión de enfermedad de Addison, enfermedad de Graves y diabetes mellitus tipo 1.1 El SPGA-II está asociado con los haplotipos HLA-DR3 y HLA-DR4 y la regla de la herencia es una forma autosómica dominante con expresividad variable.1,3
CASO CLÍNICO
Paciente de 35 años de edad, blanca, maestra de profesión, sin antecedentes familiares de interés, con diabetes mellitus tipo 1 desde hace 15 años, para lo cual recibe tratamiento con insulina novolenta y dieta; hace 9 años le fue diagnosticada una tiroiditis de Hashimoto, recibiendo tratamiento con levotiroxina sódica (100 mcg/día). Desde hace tres meses comienza a presentar adinamia marcada, anorexia, pérdida de peso, hambre de sal, náuseas, vómitos repetidos, alopecia, fatiga, trastornos visuales, somnolencia, pérdida de la memoria y estreñimiento marcado; por lo que tuvo que abandonar sus labores habituales. En el interrogatorio del aparato ginecológico se recoge el antecedente de menarquia a los 14 años, menorragias frecuentes alternando con amenorrea de casi cinco años de evolución y tres abortos espontáneos sin lograr un embarazo a pesar del estudio de infertilidad.
Examen físico: marcada afección del estado general, frialdad en las extremidades, alopecia frontal, axilar y pubiana; melanodermia de las zonas expuestas, encías, ambas manos, areolas y pezones, genitales y cicatrices. (Figura 1, 2, 3)
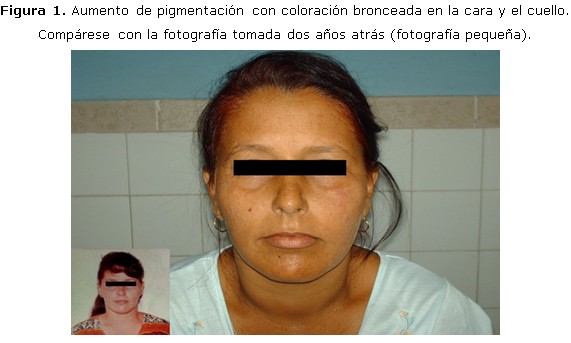
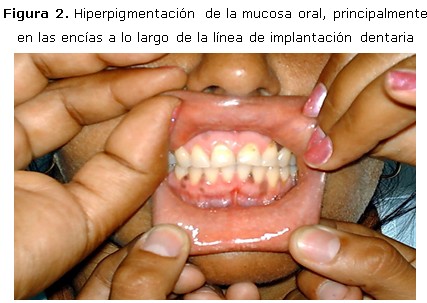
Aparato cardiorrespiratorio: murmullo vesicular normal, ruidos cardíacos rítmicos, bradicárdicos, pobremente audibles, sin soplos; tensión arterial: 80/50 mm Hg; frecuencia cardíaca: 54 lpm. Exploración neurológica: vigil, orientada en tiempo, espacio y persona, que coopera de forma lenta con bradipsiquia y bradilalia; reflejos osteotendinosos muy disminuidos, hipoacusia bilateral; sin déficit motor ni signos meníngeos. Exploración ginecológica: ambas mamas disminuidas de tamaño con areolas melánicas, pezones pequeños, caída marcada del vello pubiano, melanodermia de labios mayores y menores. Tacto vaginal y examen con espéculo: normal. Fondo de ojo: retinopatía diabética no proliferativa. Resto de la exploración: normal.
Estudios analíticos y evolución
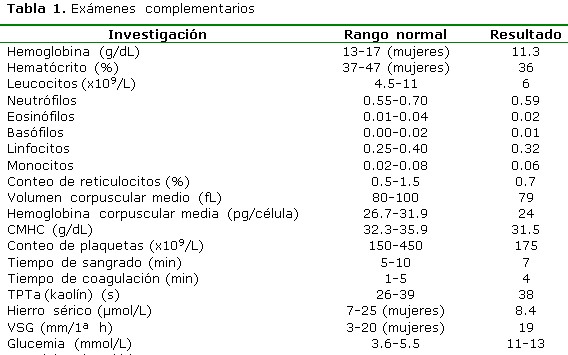
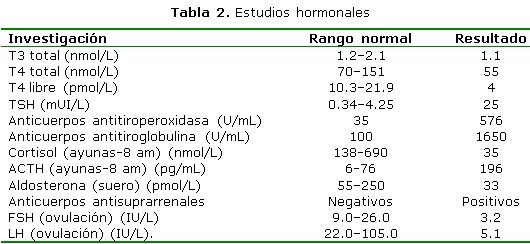
Las investigaciones de laboratorio y los estudios hormonales (la mayoría dosificados en La Habana) realizados a la paciente durante la hospitalización. Antígeno de superficie para hepatitis B, anticuerpos contra el virus de la hepatitis C, VDRL, VIH: todos normales. Prueba de Mantoux: 2 mm. Prueba de Coombs directa e indirecta, fenómeno LE, proteína C reactiva, prueba del látex para factor reumatoideo, crioglobulinas, nivel de complemento sérico, anticuerpos antinucleares: sin alteraciones. Estudio inmunogenético de HLA-B27: negativo. Densidad y sedimento urinario según conteo de Addis (2 horas): normal. Ultrasonografía (USG) de glándula tiroidea: estructura no homogénea con dimensiones de 30x24 mm y lóbulo medio prominente; no adenopatías, no calcificaciones. Citología por aspiración con aguja fina de tiroides: presencia de abundantes linfocitos, no se observan células neoplásicas. Prueba de sobrecarga de agua: positiva. (Tabla 1, 1a y 2)

Electrocardiograma: bradicardia sinusal (50 latidos por minuto) con QT prolongado. USG abdominal y transvaginal: sin alteraciones abdominales, se observan varios folículos en ambos ovarios. Mamografía bilateral: sin alteraciones. Radiografía de tórax: índice cardiotorácico normal, no alteraciones pleuro-pulmonares. Tomografía axial computarizada (TAC) de tórax y de hipófisis: sin alteraciones. Resonancia magnética nuclear de hipófisis: normal. TAC de riñones y glándulas suprarrenales: riñones normales, suprarrenales atróficas, con pequeñas zonas hipodensas centrales, no hay signos de hemorragia ni calcificaciones.
La paciente fue tratada con volumen (cloruro de sodio 0.9 %) intravenosa (IV) de forma continua, dieta rica en sodio (5 g/día), con dieta de diabético de 1 800 kcal/día. Se emplearon hidrocortisona 100 mg IV durante tres días y después 30 mg/día fraccionados (20/10); fludrocortisona 0.1 mg el primer día y después 0.05 mg/día y levotiroxina (100 µg) 1 ½ tabletas a las 9 pm; además de insulina novolenta. La paciente mejoró notablemente con la terapéutica impuesta y después de 15 días fue referida al hospital ginecológico para concluir el estudio del hipogonadismo.
DISCUSIÓN
Este síndrome es una rara poliendocrinopatía caracterizada por la enfermedad severa de múltiples glándulas endocrinas causada por trastornos inmunes con destrucción de los tejidos endocrinos.4,5 Ocurre alrededor de la tercera y cuarta décadas de la vida, y se reporta con mayor frecuencia en mujeres en relación a los hombres con una proporción de 3-4:1; en Estados Unidos se informaron aproximadamente de 14-20 personas por millón de habitantes, y se señala que esta enfermedad es mucho más frecuente si se incluyen las formas subclínicas.3 Los reportes de la morbilidad y mortalidad de esta enfermedad en el momento actual no se han podido estimar.3,4
Las otras dos endocrinopatías autoinmunes existentes son llamadas tipo I y tipo III, la primera es muy rara y propia de los niños; la tipo III se observa en asociación a enfermedades autoinmunes del tiroides junto con otras dos enfermedades inmunes, que incluyen la diabetes mellitus tipo 1, la anemia perniciosa o una enfermedad no endocrina autoinmune en ausencia de enfermedad de Addison autoinmune.1,2,4
La causa de esta enfermedad es muy poco conocida. Se señala la asociación de la diabetes mellitus tipo 1 o el hipotiroidismo a la rubéola congénita (transmisión materno-fetal del virus); por estimulación inmune de ciertas proteínas de la dieta; por una susceptibilidad genética; o por disfunción idiopática inmunopatológica.4 Estudios en ratones infectados por citomegalovirus han demostrado una enfermedad similar al síndrome poliglandular autoinmune tipo II, sin embargo no se ha reportado esta relación en humanos.3 Esta enfermedad es de naturaleza familiar pero no existe un patrón de transmisión mendeliano característico.1,3 En la enferma no se encontraron antecedentes familiares o de otro tipo.
En un estudio realizado por Dittmar y Kahaly 6 en 160 casos con síndrome poliglandular autoinmune, hallaron 151 pacientes con el tipo II, donde las enfermedades mayormente reportadas son: la diabetes mellitus tipo 1 (61 %), la tiroiditis de Hashimoto (33 %), la enfermedad de Graves (33 %), el vitíligo (20 %), la enfermedad de Addison (19 %), la alopecia (6 %), el hipogonadismo (5 %) y la anemia perniciosa (5 %); la más común de las combinaciones en esa serie fue la de diabetes mellitus tipo 1 con enfermedad de Addison y tiroiditis de Hashimoto. La paciente presentó un síndrome poliglandular dado por diabetes mellitus tipo 1, hipotiroidismo primario, enfermedad de Addison, hipogonadismo y alopecia.
El intervalo de presentación más largo entre la primera y la segunda endocrinopatía en la serie de Dittmar y Kahaly6 ocurrió entre la diabetes mellitus tipo 1 y el hipotiroidismo (13.3 +/- 11.8 años) y entre el vitíligo y las enfermedades del tiroides (16.3 +/- 13.3 años), donde el intervalo es más corto entre la enfermedad de Addison y las enfermedades del tiroides; en la paciente la primera enfermedad diagnosticada fue la diabetes mellitus a los 20 años, seguida de la tiroiditis de Hashimoto a los 26 años y la enfermedad de Addison en el momento actual, al igual que la alopecia; el hipogonadismo se observó con manifestaciones clínicas a partir de los 30 años.
La diabetes mellitus tipo 1 puede asociarse, preceder o suceder a las manifestaciones de insuficiencia tiroidea y adrenal, al igual que las manifestaciones de hipotiroidismo, que similarmente, pueden preceder o continuar a las de la insuficiencia adrenal;7 sin embargo, se señala que la diabetes mellitus tipo 1 es el componente más frecuente y el primero del síndrome poliglandular autoinmune tipo II,1, 4, 6 como ocurrió en el caso estudiado; la asociación de diabetes mellitus tipo 1 con enfermedad de Addison e hipotiroidismo primario ha recibido el epónimo de síndrome de Schmidt-Carpenter.1, 7
El síndrome poliglandular autoinmune tipo II es un complejo de enfermedades con diversas formas clínicas de presentación resultado del daño de múltiples glándulas endocrinas afectadas, como evidenciamos en nuestra paciente, con múltiples manifestaciones todas relacionadas con las insuficiencias de las glándulas comprometidas; llamando la atención la severa hiponatremia de 100 mmol/L, dato mencionado por otros autores como Gumieniak y Farwell 8 en un paciente con 101 mmol/L de sodio sérico como debut de la enfermedad.
El diagnóstico es usualmente establecido por el examen clínico y el estudio de los niveles de hormonas en suero como ACTH, cortisol, TSH, triyodotironina, tiroxina, renina en plasma, FSH, LH y electrólitos,9 además debe realizarse como complemento indispensable la detección de anticuerpos contra las diferentes glándulas endocrinas;10, 11 todos se practicaron en este caso, quedando pendiente del estudio del hipogonadismo que puede igualmente presentar un origen autoinmune, por lo regular una ooforitis o hipopituitarismo que trae consigo infertilidad y trastornos que repercuten en la esfera ginecológica e incluso cardiovascular,11, 12, 13 por lo que se requiere como mínimo una evaluación clínica completa con biopsia de ovario y anticuerpos contra células responsables de la esteroidogénesis en el ovario, entre otros.
Se debe realizar el diagnóstico diferencial con otros procesos que afectan diferentes glándulas y órganos, que tengan un origen inmune, cromosómico, hematológico o gastroenterológico, tales como: el timoma, la enfermedad celíaca, el síndrome de POEMS (polineuropatía, organomegalia, endocrinopatía, componente “M” y cambios en la piel-“Skin”), síndrome de DiGeorge (hipoparatiroidismo debido a agenesia glandular y candidiasis mucocutánea), síndrome de Kearns-Sayre (hipoparatiroidismo, hipogonadismo primario, diabetes mellitus tipo 1, y panhipopituitarismo), síndrome de Wolfram (diabetes insípida congénita y diabetes mellitus), síndrome IPEX (inmunodisregulación, poliendocrinopatía, enteropatía, herencia ligada al cromosoma X), rubéola congénita (diabetes mellitus tipo 1 e hipotiroidismo),1-5 disfunción asociada a terapéutica con interferón alfa,14 afección endocrina paraneoplásica,15 entre otros.
La terapéutica empleada en la paciente fue eficaz, por lo que se le dio el alta hospitalaria a los 15 días, refiriéndola al hospital ginecológico para concluir el estudio de su hipogonadismo.
CONCLUSIONES
Este síndrome es una rara y severa enfermedad, de incidencia desconocida en nuestro medio, con afección de múltiples glándulas endocrinas causada por trastornos inmunes con destrucción de los tejidos. El diagnóstico es fundamentalmente clínico, comprobado por la determinación de los niveles hormonales y las pruebas de inmunidad. Se debe diferenciar de otros procesos de causa inmunológica. La respuesta terapéutica generalmente es eficaz.
REFERENCIAS BIBLIOGRÁFICAS
1. Pourmotabbed G, Talavera F, Chausmer AB, Cooper M, Griffing GT, editors. Polyglandular Autoimmune Syndrome, Type II [Internet]. New York: www.emedicine.com; 2012 [cited 2011 Jun 15]. Available from: http://emedicine.medscape.com/article/124287-overview
2. Kronenberg HM. Polyglandular disorders. In: Goldman L, Schafer AI, editors. Goldman’s Cecil Medicine. 24th ed. Saunders Elsevier: Philadelphia; 2012. p. 1505-09.
3. Betterle C, Lazzarotto F, Presotto F. Autoimmune polyglandular syndrome Type 2: the tip of an iceberg?. Clin Exp Immunol. 2004 Aug;137(2):225-33.
4. Gagel RF, Jimenez C. Disorders Affecting Multiple Endocrine Systems. In: Longo DL, Kasper DL, Jameson JL, Fauci AS, Hauser SL, Loscalzo J, editors. Harrison’s Principles of Internal Medicine [CD-ROM]. 18th ed. New York: McGraw-Hill; 2012.
5. Karamifar H, Dalili S, Karamizadeh Z, Amirhakimi G, Dalili H. Autoimmune polyglandular syndrome type 2: an unusual presentation. Acta Med Iran. 2010 May-Jun;48(3):196-7.
6. Dittmar M, Kahaly GJ. Polyglandular autoimmune syndromes: immunogenetics and long-term follow-up. J Clin Endocrinol Metab. 2003 Jul;88(7):2983-92.
7. Carpenter C, Solomon N, Silverberg SG, Bledsoe T, Northcutt RC, Klinenberg JR, et al. Schmidt’s syndrome (thyroid ans adrenal insufficiency): a review of the literature and a report of fifteen new cases including ten instances of co-existent diabetes mellitus. Medicine (Baltimore). 1964 Mar;43:153-80.
8. Gumieniak MD O, Farwell MD AP. Schmidt'S syndrome and severe hyponatremia: report of an unusual case and review of the related literature. Endocr Pract. 2003 Sep-Oct;9(5):384-8.
9. Locher R, Kohler S, Schwanda S, Schmid C. Tiredness, hyperpigmentation, weight loss, nausea and vomiting. Polyglandular autoimmune syndrome (PAS) type 2. Praxis (Bern 1994). 2010 Oct 6;99(20):1223-8.
10. Bizzaro N. The predictive significance of autoantibodies in organ-specific autoimmune diseases. Clin Rev Allergy Immunol. 2008 Jun;34(3):326-31.
11. de Graaff LC, Smit JW, Radder JK. Prevalence and clinical significance of organ-specific autoantibodies in type 1 diabetes mellitus. Neth J Med. 2007 Jul-Aug;65(7):235-47.
12. Kauffman RP, Castracane VD. Premature ovarian failure associated with autoimmune polyglandular syndrome: pathophysiological mechanisms and future fertility. J Womens Health (Larchmt). 2003 Jun;12(5):513-20.
13. Wellons M. Cardiovascular Disease and Primary Ovarian Insufficiency. Semin Reprod Med. 2011 Jul;29(4):328-41.
14. Krysiak R, Boldys A, Okopien B. Autoimmune polyglandular syndrome type 2 induced by treatment with interferon alpha. Am J Med Sci. 2011 Jun;341(6):504-7.
15. Watanabe S, Tanaka J, Ohta T, Yoshizawa H, Gejyo F. Paraneoplastic neurological syndrome and polyglandular autoimmune syndrome type 2 in a case of small cell lung cancer. Thorax. 2008 Dec;63(12):1118-9.
Recibido: 6 de febrero de 2013
Aprobado: 4 de abril de 2013
Dr. Rafael Pila Pérez. Especialista de II Grado en Medicina Interna. Profesor Titular y Consultante. Camagüey, Cuba. Email: rvpila@finlay.cmw.sld.cu


{kind=link}